



Agentic AI in Pharma: From Drug Discovery to Regulatory Filing

What Is Agentic AI in Pharma?
Agentic AI in pharma refers to autonomous artificial intelligence systems designed to execute complex, multi-step workflows in drug discovery, clinical trials, and regulatory compliance with minimal human intervention. Unlike traditional machine learning models that provide static predictions (e.g., predicting toxicity), agentic AI systems proactively plan, reason, and use external tools (such as molecular simulators, database queries, and document generators) to achieve high-level goals.
For example, a drug discovery agent can autonomously identify a target, screen millions of compounds, simulate binding affinity, and propose a shortlist of candidates for synthesis.
In 2026, the life sciences industry has passed a clear inflection point. According to Menlo Ventures' State of AI in Healthcare survey of 700+ industry executives (conducted with Morning Consult, Aug–Sep 2025), 66% of pharma companies are actively building or fine-tuning proprietary AI models, and domain-specific AI adoption across healthcare has grown at 7× the rate seen in 2024, faster than any other sector surveyed. In our own work at Vector Labs across European pharma and MedTech clients, we observe the same trajectory: organisations that spent 2024 running isolated AI pilots are now building production-grade agentic workflows.
Source: Menlo Ventures, The State of AI in Healthcare, October 2025. Survey of 700+ CxO and SVP/VP executives across pharma, biotech, health systems, and payers. menlovc.com
Why Agentic AI in Pharma? Why Now?
For the past decade, the pharmaceutical industry has been data-rich but insight-poor. The sector has collectively spent billions digitising electronic lab notebooks (ELNs), sequencing genomes, and aggregating real-world evidence. Yet the cost of bringing a new drug to market remains stubbornly high — over $2 billion on average — and the timeline still drags on for 10–12 years.
Traditional AI and Machine Learning (ML) promised a revolution but delivered fragmented utility. A predictive model might tell you a molecule is toxic, but it won't tell you how to fix it. A natural language processing (NLP) tool might extract data from a clinical report, but it can't draft a regulatory submission.
This is where Agentic AI changes the game.
We are shifting from the "Oracle" paradigm (asking AI a question and getting an answer) to the "Agent" paradigm (giving AI a goal and letting it execute the work). Three converging forces are driving this shift right now:
- Regulatory Complexity. The volume of data required for FDA and EMA submissions has exploded. Human teams simply cannot manually collate and verify millions of pages of documentation without error.
- The Patent Cliff. Major blockbusters are losing exclusivity. Pharma giants need to refill pipelines faster than humanly possible.
- Generative Reasoning. Large Language Models (LLMs) have evolved from text generators to reasoning engines capable of planning and tool use.
The Five Agentic AI Architectures Transforming Pharma
Not all agents are created equal. In our work at Vector Labs across Europe, we see five distinct architectural patterns delivering value in production today.
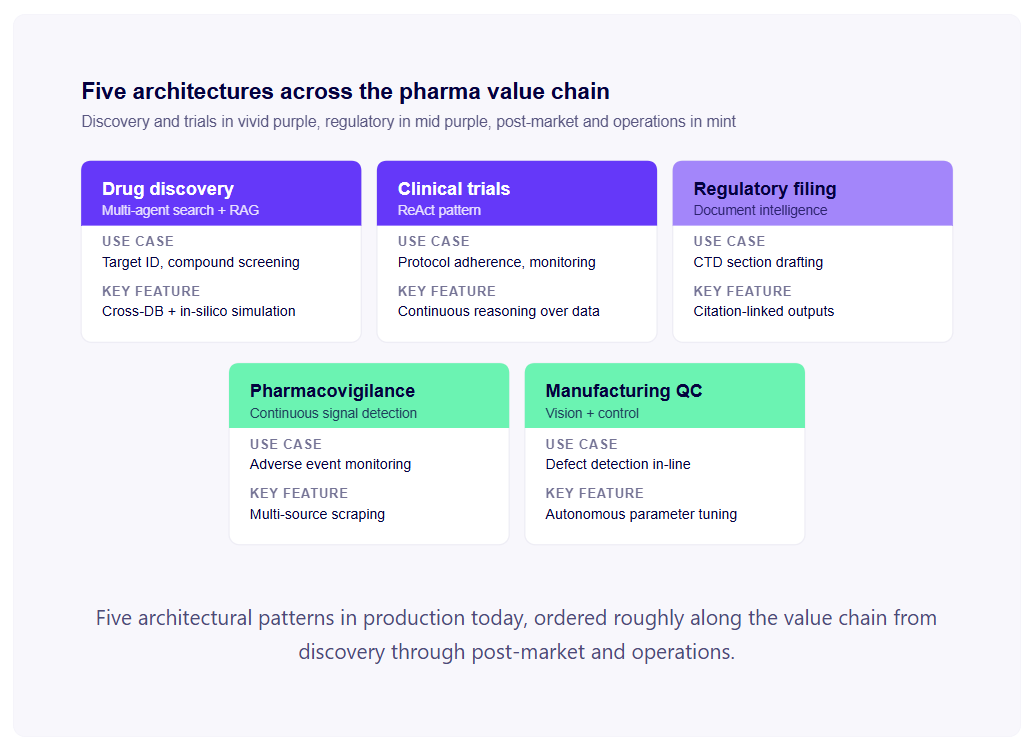 Drug Discovery Agents (Multi-Agent Search + RAG)
Drug Discovery Agents (Multi-Agent Search + RAG)
These systems use a "Planner" agent to decompose the drug design process into specialised subtasks executed by collaborating workers. A literature subagent retrieves relevant publications via RAG over PubMed, bioRxiv, and proprietary chemistry corpora. A target subagent queries protein structure databases (PDB, AlphaFold predictions) and binding-site annotations. A chemistry subagent generates candidate molecules using generative models (REINVENT, MolGAN, or transformer-based generators) and scores them against ADMET filters. A simulation subagent runs in-silico docking, binding affinity prediction, or molecular dynamics where required.
The orchestrator coordinates these workers, iterating on hypotheses, applying chemical filters, and ranking proposals by predicted potency, selectivity, drug-likeness, and synthetic accessibility. A freedom-to-operate subagent cross-references patent databases to flag IP conflicts before chemists invest in synthesis. The output is not just a list of molecules but a ranked shortlist with reasoning chains: why each candidate was selected, which targets it engages, what risks were considered.
In practice, this compresses hit-to-lead timelines from 12–18 months to 3–6 months for tractable targets. Human medicinal chemists remain in the loop for synthesis decisions, IP strategy, and proposing the "wild card" molecules that purely model-driven searches under-explore.
Clinical Trial Intelligence (ReAct Pattern)
Using the ReAct (Reason + Act) pattern, these agents continuously interleave reasoning steps with actions against trial data systems. A typical decision chain: "Patient X has elevated ALT and AST → this exceeds the protocol's hepatic exclusion threshold (criterion 4.2) → query the prior visit data → confirm the trend is not isolated → flag to the medical monitor with severity classification → draft a preliminary safety narrative for review."
Practical deployments span several workflows. Patient recruitment agents scan unstructured EHR notes across partner hospitals, matching candidates to complex eligibility criteria with linked-source reasoning so the medical monitor can verify each match. Protocol deviation detection agents monitor visit data and flag departures from the planned schedule, dosing, or assessment windows. Safety triage agents pre-classify incoming reports by seriousness and expectedness ahead of human review. DSUR/PSUR drafting agents assemble the periodic safety reports from accumulated case data.
The regulatory bar is high. Every reasoning step must be loggable, every action against the data must be auditable, and human medical monitors retain decision authority on safety calls. The economic case is in the time savings: trial recruitment cycles shortened by 30–40%, protocol deviation review compressed from weekly to continuous, and DSUR drafting time cut by half.
Regulatory Filing Agents (Document Intelligence)
These agents are "writers" with strict fact-checking constraints. They ingest CSRs, statistical analysis plans, raw SAS outputs, and study protocols, then draft sections of the Common Technical Document (CTD) — Module 2 summaries (Clinical Overview, Summary of Clinical Safety, Summary of Clinical Efficacy), Module 5 study reports, and the narrative sections that bridge structured data to regulatory prose.
The "fact-checking constraint" is the architectural distinction. Every sentence the agent writes must be traceable to a source — a specific table, a paragraph in a CSR, a statistical output. A validator subagent independently checks that citations resolve to the claimed source and that the numbers in the generated text match the source data exactly. Hallucinated numbers or unsupported claims block at the validator gate; nothing reaches the human reviewer without source linkage.
The medical writer workflow shifts from drafting to reviewing. Where the writer previously wrote a 200-page Clinical Overview from scratch, they now edit, refine, and verify a structured draft assembled by the agent in hours rather than weeks. We've measured medical writer capacity gains of 2–4× on suitable document types, with the time saved redirected to higher-value work: argumentation strategy, regulatory positioning, and response to FDA/EMA queries.
Pharmacovigilance Agents
Continuous monitoring agents that aggregate signals across heterogeneous sources — published medical literature, patient forums and social media, internal safety databases (Argus, ArisGlobal), regulatory adverse event databases (FAERS, EudraVigilance, WHO VigiBase), and call-centre transcripts. The agent normalises mentions into structured Individual Case Safety Reports (ICSRs) compatible with E2B(R3) data exchange, classifies seriousness and expectedness, and surfaces emerging signals for human pharmacovigilance review.
The economic case is partly speed and partly coverage. Manual triage of incoming case reports is one of the most labour-intensive activities in pharmacovigilance — agents can process incoming reports in minutes rather than the hours of manual triage per case, reallocating safety physicians to genuine signal evaluation rather than data entry. On the coverage side, an agent reviewing thousands of patient-forum posts a week can surface adverse-event signals that wouldn't reach the formal ICSR pipeline for months.
The regulatory expectations are strict. Pharmacovigilance is one of the most regulated functions in pharma, with reporting timelines of 15 days for serious unexpected ADRs and 90 days for non-serious. Agents must produce auditable case files with verified source citations, and human pharmacovigilance physicians retain final classification authority. Implementations that bypass human review on case classification create regulatory risk that's not worth the efficiency gain.
Manufacturing Quality Control
Vision-based agents in the factory operating under Pharma 4.0 and Process Analytical Technology (PAT) frameworks. The agents combine real-time imaging (tablet inspection, vial fill verification, capsule defect detection) with closed-loop control over upstream machinery — adjusting press force, fill speed, or coating parameters before defects propagate into rejected batches.
The shift is from reactive quality control (inspect after the fact, reject defects) to predictive process control (detect drift in process parameters before defects occur). A tablet press exhibiting slight weight variation can be re-tuned in real time rather than producing a batch that must later be scrapped. A vial filling line drifting toward over-fill can be corrected mid-run rather than after QA sampling identifies the issue hours later.
Economic impact is direct: pharma batch rejections can carry six- and seven-figure costs each, and yield improvements of 1–3% on high-value biologics translate to material revenue. The regulatory framing requires careful design, closed-loop control changes within validated GMP processes need predetermined boundaries, documented change-management protocols, and alignment with the validated Master Batch Record. Done correctly, the agent operates as an extension of the validated process, not a circumvention of it.
How Do You Measure Economic Impact in Pharma AI?
Too many pharma AI projects die in "Pilot Purgatory" because they lack a clear economic framework. At Vector Labs, we advise CTOs and CDOs to measure impact across four dimensions.
Time compression. Reducing cycle time on drug discovery, trial recruitment, regulatory documentation, or pharmacovigilance review. Measured as days or months saved per workflow — and in pharma, time is money in a direct sense: every day of clinical trial delay carries quantifiable cost.
Direct cost reduction. Operational savings from automation — reallocated FTE hours, reduced rework, fewer external consulting cycles, lower CRO spend on tasks now executed internally.
Quality and compliance lift. Reduced regulatory queries, fewer GxP findings, lower submission rework rates, fewer pharmacovigilance signals missed. Often the largest hidden value driver in regulated environments — a deficiency letter that delays approval by six months has costs that dwarf the AI program.
Strategic optionality. New capabilities the agent enables — pursuing therapeutic areas previously deprioritised due to cost, faster trial iteration, faster response to safety signals, the ability to participate in indications that were previously economically unviable. Hardest to quantify, most transformative when realised.
Cost Structure: Traditional vs. Agentic
The cost profile of agentic AI is structurally different from traditional pharma software. Traditional pipeline software carries high upfront licensing with low marginal cost. Agentic AI carries moderate upfront integration cost plus variable inference cost (LLM tokens, compute) that scales with usage. For pharma operations where the alternative is FTE hours at €100–300 per hour, agentic AI economics become favourable even at meaningful token usage — and the marginal cost of the next workflow execution is typically near-zero.
What's Actually Working in 2026: Three Real Implementations
Case A: The "Fail Fast" Drug Discovery Agent
A mid-sized biotech deployed a multi-agent system to screen kinase inhibitors. The system didn't just find hits; it autonomously cross-referenced patent databases to ensure freedom-to-operate. Result: they identified a lead candidate in 4 months instead of 18, with a 10× reduction in synthesis costs.
Case B: The Clinical Trial Matchmaker
A global pharma company used a ReAct-based agent to scan unstructured electronic health records (EHRs) across partner hospitals. The agent matched patients to complex oncology protocols with 95% accuracy. Result: recruitment timeline reduced by 40%, saving an estimated €25K per day in delayed trial costs.
Case C: Automated Regulatory Submission (Vector Labs)
We built a "Regulatory Co-pilot" for a UK-based pharma client. The system ingests raw SAS outputs and study reports, then drafts the narrative sections of the Clinical Study Report. Result: medical writers shifted from drafting to reviewing, increasing their capacity by 3×.
The Three Mistakes Pharma Companies Make with Agentic AI
Mistake 1: Treating It Like Traditional ML
Leaders ask, "What is the accuracy of this model?" But agents aren't classifiers; they are workers. The right metric is "success rate of task completion," not just F1-score.
Mistake 2: Ignoring Regulatory Architecture Requirements
You cannot use a "black box" agent in GxP environments. If an agent makes a decision, it must be traceable. We see companies build impressive demos that are not legally deployable because they lack deterministic audit trails.
Mistake 3: Underestimating Data Quality Needs
An agent is only as good as the tools and data you give it. If your internal data is trapped in scanned PDFs or siloed legacy systems, the agent will hallucinate or fail. Data engineering is 80% of the battle.
Architecture Considerations for Regulated Industries
Building for pharma requires a different architectural standard than building a consumer chatbot.
Explainability (XAI). Compliance with 21 CFR Part 11, EU Annex 11, and GxP standards means every output must be explainable. We use "Chain-of-Thought" logging where the agent writes down its reasoning before acting.
Deterministic Guardrails. Agents should have creative freedom in search but strict constraints in output. We use validator agents to check every output against a "truth file."
Human-in-the-Loop (HITL). The agent prepares the decision; the human signs it off. This preserves the legal chain of custody for liability.
Data integrity (ALCOA+). Every action the agent takes must be Attributable, Legible, Contemporaneous, Original, Accurate — plus Complete, Consistent, Enduring, and Available. Building these properties into the agent's logging and storage architecture from day one is significantly cheaper than retrofitting them.
The Build vs Buy Decision for Pharma AI
Three patterns, with different risk profiles.
Build internally. Right when your internal data and workflows are highly specific, your team has ML engineering depth, and the agent's workflow is competitively differentiating. Timeline: 12–24 months to production. Cost: €500K–€2M+ depending on scope. The advantage is full control over data, IP, and integration. The risk is execution.
Buy a vertical platform. Right when a vendor solves your specific workflow — regulatory drafting, pharmacovigilance, trial matching — with existing GxP-aligned compliance and validated outputs. Faster time-to-value (3–6 months). Limited customisation and licensing dependencies. The advantage is speed. The risk is vendor lock-in and inability to differentiate.
Hybrid: partner and extend. The most common pattern in 2026. License a vertical platform for the core workflow, build extensions for your specific data, internal integrations, and competitive use cases. Pairs vendor speed with internal differentiation. The advantage is balance. The risk is integration complexity across two stacks.
Conclusion: What Pharma Leaders Should Do Next
The era of AI as a science experiment is over. In 2026, the question is not "Can AI work?" but "How fast can we integrate agents into our regulated workflows?"
To move forward, I recommend three immediate actions:
- Audit your data readiness. Can an agent access your historical trial data via API?
- Pick one "vertical slice." Don't try to solve "Drug Discovery." Solve "Hit-to-Lead Optimization" or "CSR Drafting."
- Partner with specialists who understand GxP. Generalist AI firms often crash on the rocks of pharmaceutical regulation.
FAQs
Agentic AI refers to autonomous systems that can pursue complex goals (such as designing a molecule or drafting a report) by planning workflows, using tools, and iterating on feedback. Unlike passive ML models, agents take action to achieve outcomes.
A pilot typically takes 3–4 months to reach MVP (Minimum Viable Product). Full enterprise deployment, including validation and integration with legacy systems, usually takes 6–12 months depending on data readiness.
ROI is significant. Companies often see a 10× increase in screening capacity and a 30–50% reduction in early-stage discovery timelines. The strategic ROI comes from "failing fast": avoiding expensive clinical failures.
Yes, provided they are architected correctly. Compliance requires features like audit logs, version control, deterministic outputs, and human-in-the-loop validation workflows to satisfy 21 CFR Part 11, EU Annex 11, and GxP standards.
No. Agents replace the drudgery (data entry, literature review, initial screening), allowing scientists to focus on high-value strategy, hypothesis generation, and complex decision-making.
Agents need access to structured data (databases, registries) and unstructured data (PDFs, reports). High-quality, curated internal data is more valuable than massive public datasets for building competitive advantage.
Costs range from €150K–€400K for specific workflow pilots to millions for end-to-end platforms. However, the cost is often negligible compared to the daily cost of clinical trial delays or lost exclusivity.
Traditional ML is predictive (e.g., "This molecule is toxic"). Agentic AI is proactive and executive (e.g., "This molecule is toxic, so I have designed 5 alternatives and simulated their safety profiles for your review").