



Clinical validation of AI diagnostics: what the evidence actually needs to show

What clinical validation for AI medical devices actually requires under EU MDR and FDA: pre-specification, multi-site evidence, reader studies, and the gap between what most teams have done and what regulators expect.
"Validated" is the most overused word in health AI. Every vendor has a validated algorithm. Every pitch deck references validation studies. Almost none of them mean the same thing, and most of them don't mean what a notified body or FDA reviewer means when they ask for clinical validation evidence.
This article is about what clinical validation for AI diagnostics actually requires. Specifically, what it needs to show, how it needs to be structured, and what the most common gaps are between what founders believe they've done and what regulators require.
What regulators mean by clinical validation
Clinical validation for AI medical devices has a specific technical meaning that is different from common usage.
In the software engineering context, "validation" means confirming that you built the right thing — that the software meets user requirements. In the medical device regulatory context, clinical validation means demonstrating that your device achieves its intended clinical purpose in its intended patient population, used as intended, by intended users, in the intended clinical environment.
This is a much higher bar than "our algorithm achieved 95% accuracy on a held-out test set."
EU MDR requires a clinical evaluation — a systematic and planned process to continuously generate, collect, analyse, and assess clinical data pertaining to the device in order to verify its safety and performance throughout its intended lifetime. The clinical evaluation is documented in a Clinical Evaluation Report (CER), structured in line with MDCG 2020-1 (the EU's guidance on clinical evaluation of medical device software).
FDA requires valid scientific evidence — evidence from well-designed studies conducted and reported in accordance with generally recognised scientific principles, sufficient to support a determination that there is reasonable assurance the device is safe and effective under its conditions of use.
Both frameworks converge on the same underlying requirement: rigorous, pre-specified, independent evidence that your device works as claimed in real clinical conditions. Test set accuracy is a component of this evidence. It is not sufficient on its own.
The evidence hierarchy
Not all clinical evidence is equal. Both EU MDR and FDA use an implicit evidence hierarchy when assessing the strength of a clinical validation package.
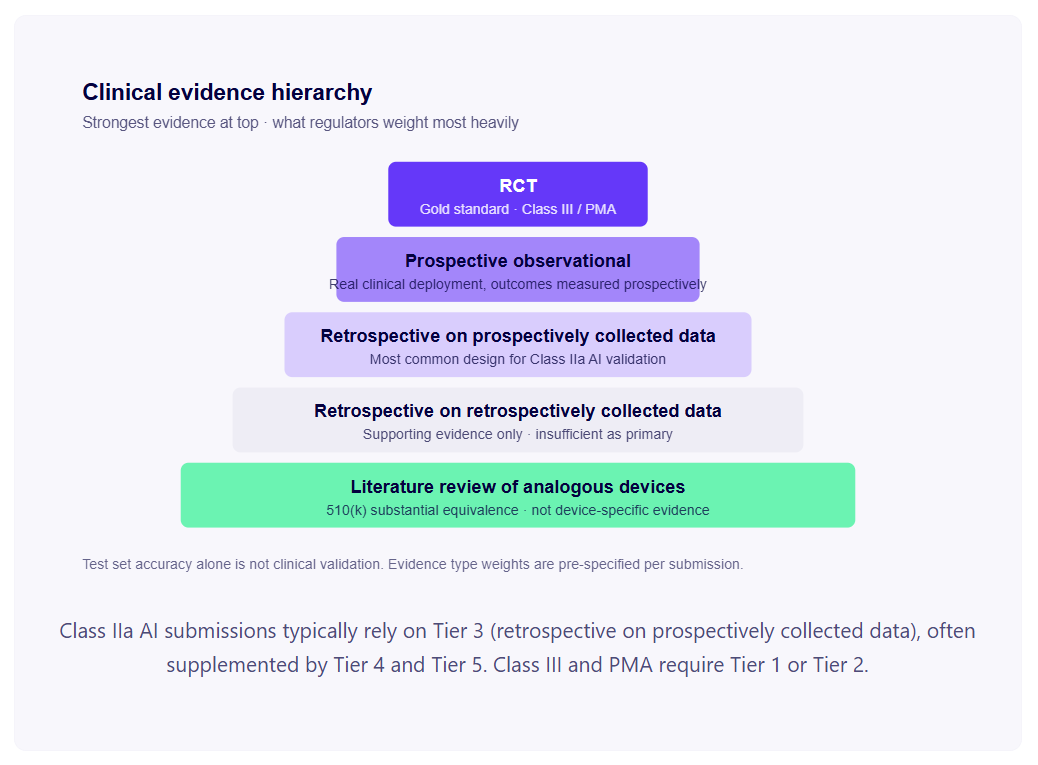
Prospective randomised controlled trials (RCTs) are the gold standard, required for Class III / PMA devices. Rarely required for Class IIa diagnostic decision-support tools — but the absence of any prospective data is increasingly being questioned by notified bodies for high-impact diagnostics.
Prospective observational studies deploy your algorithm in a real clinical environment and measure outcomes prospectively. Strong evidence for diagnostic performance in conditions of actual clinical use.
Retrospective studies on prospectively collected data are the most common design for AI diagnostic validation. A dataset of clinical cases collected prospectively (for example, routine clinical imaging with confirmed diagnoses from standard care pathways) is used retrospectively to evaluate algorithm performance. Acceptable when the prospective collection followed good clinical practice and the algorithm had no influence on data collection.
Retrospective studies on retrospectively collected data are the weakest form of clinical evidence accepted by regulators — data collected for other purposes, cleaned and labelled for validation. Acceptable as supporting evidence but typically insufficient as primary evidence for Class IIa and above.
Literature review of analogous devices is used in 510(k) substantial equivalence arguments — published performance data from predicate devices can form part of the clinical evidence base. Not a substitute for device-specific validation in your intended patient population.
The pre-specification requirement
The single most important principle in clinical validation that most founders underestimate is pre-specification: your performance metrics, your statistical analysis plan, and your success criteria must be defined before you look at the test data.
Post-hoc metric selection — choosing the metrics that make your algorithm look best after seeing the test results — is a form of p-hacking. Regulators know it. Notified bodies know it. It invalidates the statistical inference you're trying to draw.
A pre-specified validation study defines, before analysis:
- Primary performance metrics (sensitivity, specificity, AUC, PPV, NPV — and which is primary)
- Secondary metrics and subgroup analyses
- The statistical confidence intervals or significance thresholds you'll use
- The minimum acceptable performance threshold — what performance level must the device achieve to be considered validated?
- The statistical analysis plan: how missing data is handled, how confidence intervals are calculated, which statistical tests are used
The minimum acceptable performance threshold deserves particular attention. "As good as the predicate" is not sufficient if you're claiming clinical benefit. "Clinically non-inferior to [benchmark]" requires defining the non-inferiority margin. The performance threshold should be clinically meaningful — not just statistically significant — and justified by clinical evidence about what level of performance is adequate for safe clinical use.
If your validation study was conducted without pre-specification, this doesn't automatically disqualify the results — but it substantially weakens the evidence, and you'll need to be transparent about this in your clinical evaluation report. More importantly, plan any further validation studies with pre-specification from the start.
Dataset requirements: what notified bodies and FDA actually look for
Population representativeness
Your validation dataset must include patients representative of your intended use population. "Intended use population" means the patients your algorithm will be applied to in clinical practice — which may be broader than the patient population in the hospital where you collected your training data.
If your cardiac AI was trained and validated on patients from a large urban teaching hospital in Western Europe, and you intend to deploy it across diverse European clinical settings, your validation dataset should reflect that diversity — across age, sex, ethnicity, comorbidity profile, and acquisition conditions (scanner type, image quality, clinical workflow variation).
Notified bodies increasingly expect to see statistical analysis of performance across relevant subgroups — not just aggregate performance. If your algorithm performs at 94% sensitivity on White European patients and 81% sensitivity on South Asian patients, that performance disparity is a regulatory question, not just a fairness question.
Independence of training and validation data
This is the most technically critical data requirement and the one most frequently violated in practice. Your validation dataset must be completely independent of your training dataset — not just at the case level, but at the patient level. If Patient A has three scans in your dataset, and one scan is in training and two are in validation, Patient A's data has leaked into your validation. The model may have learned patient-specific patterns that appear as generalisation in validation but would not generalise to new patients.
For retrospective studies on hospital datasets, enforcing patient-level independence requires explicit patient ID management in your data pipeline. If you didn't do this rigorously, your reported validation performance is optimistic by an unknown amount.
Consecutive or randomly sampled cases
Selection bias in your validation dataset is a significant validity threat. Selecting "interesting" or "clear" cases produces performance estimates that don't reflect real-world case mix. Consecutive case sampling (all cases meeting inclusion criteria over a defined period) or properly randomised sampling is required for a defensible validation study.
Ground truth quality
Your algorithm's performance can only be measured relative to a ground truth. The quality of your validation evidence is bounded by the quality of your ground truth labels.
For imaging diagnostics, ground truth is typically expert radiologist annotation — but expert annotation has inter-rater variability. The question regulators ask: how was annotation disagreement handled? If a case was ambiguous, what was the adjudication process? Was there a reference standard (biopsy, follow-up outcome, panel consensus) for ambiguous cases?
For predictive models (readmission prediction, disease onset prediction), ground truth comes from clinical outcomes in the records. The question: were outcomes recorded consistently and completely? Was there differential follow-up between patient groups — some patients more likely to be followed up than others — that could bias the outcome data?
Ground truth methodology needs to be documented in your study protocol and reported in your clinical evaluation. If it's weak, your performance estimates are unreliable and a notified body reviewer with clinical expertise will notice.
The multi-site requirement
Notified bodies for Class IIa AI diagnostics are increasingly expecting multi-site validation. This means performance evidence from at least two to three independent clinical sites, not just the site where the algorithm was developed.
The rationale is clear: AI models trained on data from one clinical environment often perform significantly worse in other environments due to different scanner manufacturers, models, or acquisition protocols; different patient population demographics and case mix; different clinical workflow and data recording practices; different image preprocessing pipelines.
Single-site validation performance is the best-case scenario for deployment performance. Multi-site validation is a more realistic estimate. The gap between the two is real and sometimes large — published literature documents 10–20 percentage point performance drops when diagnostic AI systems are evaluated at external sites.
Single-site validation is the most common preventable gap we see in pre-submission reviews. It generates more notified body queries than any other clinical evaluation issue — and the queries are usually fatal to the original timeline.
For a startup without multi-site data at the time of initial CE marking: be honest about this limitation in your clinical evaluation report, propose a PMCF study that will collect multi-site post-market evidence, and discuss this proactively with your notified body during pre-submission. Omitting the limitation is worse than acknowledging it.
Reader studies and clinical utility
For AI tools intended to augment clinicians like radiology decision support, ECG interpretation, pathology second-reader, the strongest evidence isn't just diagnostic accuracy. It's clinical utility: does the AI tool actually change clinician decision-making for the better?
The gold standard study design here is the reader study: a panel of clinical readers (typically 8–15) reads a fixed set of cases twice — once without the AI tool, once with it. Paired-comparison analysis measures whether AI-assisted reads are more accurate, faster, or more consistent than unassisted reads. The result quantifies the actual clinical benefit your tool delivers when used as intended.
Reader studies are substantial undertakings — 6–12 months from protocol to final report, costs in the €100K–€400K range depending on reader compensation and case volume. But they produce the most defensible evidence of clinical benefit available, and FDA increasingly requests them for AI tools intended to augment radiologists or other clinical readers. Notified bodies expect them for higher-acuity diagnostics, particularly under MDCG 2020-1.
If a reader study is impractical for initial CE marking, document why, plan one as PMCF, and be transparent about the gap.
Intended use vs. actual use: the mismatch problem
Your clinical validation is specific to your intended use i.e. the specific patient population, clinical setting, and use context documented in your intended purpose statement and your IFU.
What happens when clinicians use your algorithm outside that intended use? A diagnostic tool validated for screening adult patients gets used for paediatric patients. An algorithm validated on images from one scanner manufacturer gets used on images from another. An algorithm trained on data from secondary care gets deployed in primary care.
From a regulatory standpoint, use outside the intended use is the user's or deploying institution's responsibility, not the manufacturer's — provided your IFU clearly specifies the intended use and its limitations, your training is adequate, and your post-market surveillance captures off-label use signals.
From a clinical safety standpoint, it's your problem anyway, because your algorithm's performance outside its validated scope is unknown — and if it produces patient harm, the reputational and commercial consequences fall on you regardless of the regulatory allocation of responsibility.
The practical response is two-fold: design your intended use scope to be as clinically realistic as possible (don't artificially constrain it to make validation easier if the clinical use will be broader), and build post-market surveillance mechanisms that detect when your device is being used outside its intended scope.
The EU AI Act layer for validation
For EU-bound programs, the AI Act sits on top of MDR's clinical evaluation requirements and adds specific obligations that overlap with — but are not identical to — what notified bodies have historically expected.
The most consequential additions for clinical validation:
Training data governance — the AI Act requires documented evidence of data quality, representativeness, and bias testing in the training set, not just the validation set. This effectively extends "validation evidence" backwards into how you constructed the training data in the first place. Documentation of dataset composition, labelling methodology, and bias mitigation needs to be defensible in the AI Act conformity assessment.
Transparency to deployers — your clinical evaluation needs to provide deployers (hospitals, clinical departments) with enough performance information to operate the device safely. This typically means subgroup performance breakdowns, calibration analysis, and documented limitations, all in the deployer-facing IFU.
Human oversight provisions — for AI systems where outputs are reviewed by clinicians, the AI Act requires documented mechanisms for clinicians to override, interpret, or refuse the AI's output. The clinical evaluation needs to address how human oversight is preserved in real clinical use.
For programs already through MDR conformity assessment, AI Act compliance integrates relatively smoothly. For new entrants, plan AI Act documentation alongside the MDR clinical evaluation from day one — retrofitting is significantly more expensive than building both in parallel.
PMCF: the validation that continues after launch
Post-market clinical follow-up (PMCF) is required for Class IIa devices under EU MDR — your clinical evaluation extends beyond initial CE marking and continues throughout the device's marketed life.
PMCF is the regulatory mechanism that captures real-world performance data — performance across diverse clinical settings, long-term reliability, performance on subgroups under-represented in pre-market evidence, and detection of distributional shift. For AI products specifically, PMCF interacts with PCCP (Predetermined Change Control Plan) design: data gathered through PMCF can support retraining decisions, and PCCP-enabled model updates can be validated through PMCF infrastructure.
A credible PMCF plan in your initial clinical evaluation strengthens the submission — it shows the notified body you understand validation as an ongoing obligation, not a one-time exercise. Specific PMCF activities to plan: prospective performance monitoring at deployment sites, registry participation where relevant, planned subgroup-specific evidence collection, and explicit triggers for additional clinical investigations if monitoring signals warrant them.
For startups, the practical implication is that post-market infrastructure — performance monitoring, complaint handling, vigilance reporting, PMCF data collection — needs to be designed before clearance, not bolted on after. Notified bodies probe the PMCF plan during review; a thin plan is a red flag.
What good validation documentation looks like
The clinical evaluation report that satisfies a notified body review for a Class IIa AI diagnostic typically includes:
- Intended purpose statement — precise, complete, reviewed by a clinical expert
- Clinical background — systematic literature review of the clinical condition, existing diagnostic methods, and published AI performance data
- Clinical evidence appraisal — for each piece of clinical data, an assessment of its relevance, quality, and contribution to the evidence package
- Primary performance data — your prospective or retrospective validation study, with pre-specified metrics, confidence intervals, subgroup analyses, and comparison to benchmark
- State of the art analysis — how your device performs relative to existing diagnostic methods and comparable AI systems
- Benefit-risk analysis — the clinical conclusion: does the evidence support that the benefit of deploying this device outweighs the residual risks?
- PMCF plan — how you will continue to collect and analyse clinical data post-market
The clinical evaluator who signs this document must be a qualified clinician with relevant expertise — either a senior clinician on your team or an external clinical expert engaged for this purpose. The notified body will assess the clinical evaluator's qualifications as part of the review.
What separates a clinical evaluation that passes from one that doesn't isn't usually the accuracy of the model — it's the discipline of pre-specification, the rigour of population analysis, the honesty about limitations, and the credibility of the PMCF plan. Programs that resource clinical writing as carefully as ML engineering ship; programs that don't, don't.
FAQs
There's no universal number. It depends on your condition prevalence, primary metric, and the confidence intervals you're claiming. For a binary classification task aiming to demonstrate sensitivity 90%+ and specificity 85%+ with reasonable 95% confidence intervals, you typically need at least 50 positive cases and 200 negative cases per condition. For rare conditions (STEMI, complete heart block), targeted enrichment to reach those numbers is required. Pre-specify the sample size with a statistical power calculation in your study protocol — notified bodies probe this.
Most Class IIa AI submissions are based on retrospective studies on prospectively collected data, which is acceptable when the prospective collection followed good clinical practice and the algorithm did not influence data collection. Plan a prospective PMCF study to run post-launch, document it in your clinical evaluation, and discuss the prospective gap proactively with your notified body during pre-submission. Omitting prospective evidence entirely is increasingly questioned by notified bodies for high-impact diagnostics.
The underlying expectations have converged. Both require pre-specified, independent, rigorously analysed evidence of clinical performance in the intended population. The differences are procedural and formatting: FDA is more prescriptive about study design for novel device types (especially De Novo), uses Pre-Submission interaction to refine evidence plans, and requires the evidence in FDA's specific format. EU MDR's CER follows MDCG 2020-1's structure. Clinical data can largely be shared between submissions; submission format is built independently.
For internal benchmarking, yes. For regulatory validation, generally no — public datasets are typically used for training or development, and using them for validation creates ambiguity about independence. Your validation dataset should be properly independent of training data and ideally collected specifically for validation. Public datasets can supplement as comparison points to published performance of analogous devices.
A reader study compares clinical reader performance with and without your AI tool. It's the gold standard for demonstrating clinical utility not just diagnostic accuracy, but actual impact on clinician decision-making. FDA increasingly requests reader studies for AI tools intended to augment radiologists or other clinicians. Notified bodies expect them for higher-acuity diagnostics. They're substantial undertakings, typically 12+ readers, paired-comparison analysis, 6–12 months of execution, but produce the strongest evidence of clinical benefit available.
Document the methodology and report the disagreement rates. Common mitigations include multi-rater consensus on training labels, separate adjudicated test sets where ground truth is clinically confirmed (downstream pathology, biopsy, outcome data), and panel review of ambiguous cases. Inter-rater kappa scores should be reported in the clinical evaluation. Treating ground truth as noisy and reporting it transparently is more defensible than pretending it isn't.
Notified bodies don't set absolute thresholds. Instead they assess whether your performance is clinically meaningful relative to existing standard of care and consistent with your intended use. For STEMI flagging, sensitivities above 90% with specificities above 85% are typical published targets. For atrial fibrillation detection in screening contexts, sensitivity 95%+ at specificity 95%+. Your clinical evaluation should justify the chosen targets against published benchmarks, not present them in a vacuum.
Partially. PMCF is required for Class IIa devices to continue gathering evidence post-launch and can fill specific gaps — real-world performance across diverse sites, long-term performance, rare event detection. But PMCF can't substitute for the initial validation evidence required for clearance. Your pre-submission evidence package needs to be defensible at submission; PMCF supplements it post-launch, it doesn't replace it.