



Клинично валидиране на диагностиката с изкуствен интелект: какво всъщност трябва да покажат доказателствата

Какво всъщност изисква клиничното валидиране за медицински устройства с изкуствен интелект съгласно EU MDR и FDA: предварителна спецификация, доказателства от множество центрове, проучвания на читатели и разликата между това, което повечето екипи са направили, и това, което регулаторните органи очакват.
„Валидиран“ е най-често използваната дума в областта на здравния изкуствен интелект. Всеки доставчик има валидиран алгоритъм. Всяка презентация се позовава на валидационни проучвания. Почти никое от тях не означава едно и също нещо и повечето от тях не означават това, което нотифициран орган или рецензент на FDA имат предвид, когато искат доказателства за клинична валидация.
Тази статия е за това какво всъщност изисква клиничното валидиране за диагностика с изкуствен интелект. По-конкретно, какво трябва да показва, как трябва да бъде структурирано и какви са най-често срещаните разлики между това, което основателите смятат, че са направили, и това, което изискват регулаторните органи.
Какво имат предвид регулаторите под клинично валидиране
Клиничното валидиране на медицински устройства с изкуствен интелект има специфично техническо значение, което е различно от общоприетото използване.
В контекста на софтуерното инженерство „валидиране“ означава потвърждение, че сте създали правилното нещо – че софтуерът отговаря на потребителските изисквания. В контекста на регулаторните мерки за медицински изделия, клиничното валидиране означава демонстриране, че вашето устройство постига предвиденото си клинично предназначение в целевата популация от пациенти, използвано по предназначение, от предвидените потребители, в предвидената клинична среда.
Това е много по-висока летва от „нашият алгоритъм постигна 95% точност при дълъг тестов набор“.
Регламентът на ЕС за медицински изделия (MDR) изисква клинична оценка — систематичен и планиран процес за непрекъснато генериране, събиране, анализ и оценка на клинични данни, отнасящи се до изделието, с цел да се провери неговата безопасност и ефективност през целия му предвиден жизнен цикъл. Клиничната оценка се документира в Доклад за клинична оценка (CER), структуриран в съответствие с MDCG 2020-1 (насоки на ЕС за клинична оценка на софтуера на медицинските изделия).
FDA изисква валидни научни доказателства — доказателства от добре разработени проучвания, проведени и докладвани в съответствие с общопризнати научни принципи, достатъчни, за да подкрепят установяването, че има разумна гаранция, че устройството е безопасно и ефективно при условията на употреба.
И двете рамки се обединяват около едно и също основно изискване: строги, предварително определени, независими доказателства, че вашето устройство работи както е заявено в реални клинични условия. Точността на тестовия набор е компонент от тези доказателства. Тя не е достатъчна сама по себе си.
Йерархията на доказателствата
Не всички клинични доказателства са еднакви. Както EU MDR, така и FDA използват имплицитна йерархия на доказателствата, когато оценяват силата на пакета за клинично валидиране.
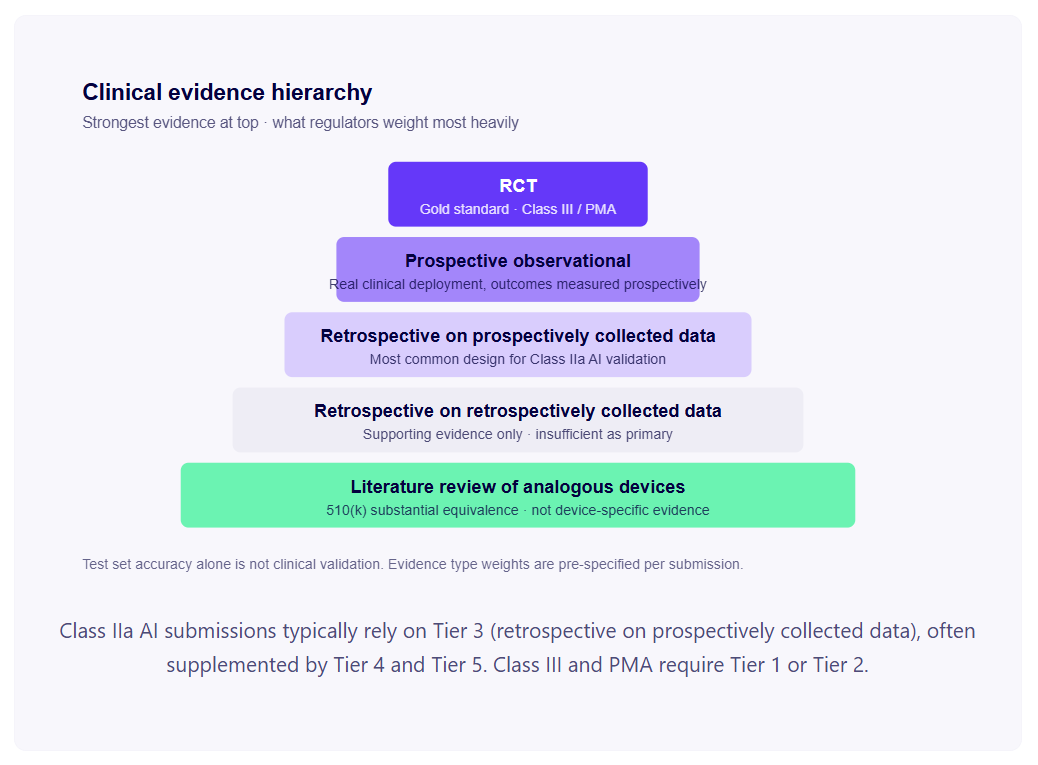
Проспективните рандомизирани контролирани проучвания (РКИ) са златният стандарт, изискван за устройства от клас III / PMA. Рядко се изискват за инструменти за подпомагане на диагностичните решения от клас IIa, но липсата на каквито и да било проспективни данни все по-често се поставя под въпрос от нотифицираните органи за диагностика с високо въздействие.
Проспективни обсервационни проучвания внедряват вашия алгоритъм в реална клинична среда и измерват резултатите проспективно. Убедителни доказателства за диагностична ефективност в условия на действителна клинична употреба.
Ретроспективните проучвания върху проспективно събрани данни са най-често срещаният дизайн за валидиране на диагностика с изкуствен интелект. Набор от клинични случаи, събрани проспективно (например рутинна клинична образна диагностика с потвърдени диагнози от стандартни пътища на лечение), се използва ретроспективно за оценка на производителността на алгоритъма. Приемливо е, когато проспективното събиране е следвало добрата клинична практика и алгоритъмът не е оказал влияние върху събирането на данни.
Ретроспективните проучвания върху ретроспективно събрани данни са най-слабата форма на клинични доказателства, приемана от регулаторните органи — данни, събрани за други цели, пречистени и етикетирани за валидиране. Приемливи като подкрепящи доказателства, но обикновено недостатъчни като първични доказателства за Клас IIa и по-високи.
Прегледът на литературата за аналогични устройства се използва в аргументи за съществена еквивалентност по 510(k) — публикуваните данни за ефективността на предикатните устройства могат да формират част от клиничната доказателствена база. Не е заместител на валидирането на специфичното за устройството място във вашата целева популация пациенти.
Изискването за предварителна спецификация
Най-важният принцип в клиничната валидация, който повечето основатели подценяват, е предварителната спецификация : вашите показатели за ефективност, вашият план за статистически анализ и вашите критерии за успех трябва да бъдат дефинирани, преди да разгледате тестовите данни.
Изборът на показатели „post-hoc“ – избирането на показателите, които правят алгоритъма ви да изглежда най-добре, след като видите резултатите от тестовете – е форма на p-хакерство. Регулаторните органи го знаят. Нотифицираните органи го знаят. Това обезсилва статистическия извод, който се опитвате да направите.
Предварително определено валидационно проучване определя, преди анализа:
- Основни показатели за ефективност (чувствителност, специфичност, AUC, PPV, NPV — и кой е първичен)
- Вторични показатели и анализи на подгрупи
- Статистическите доверителни интервали или прагове на значимост, които ще използвате
- Минималният приемлив праг на производителност — какво ниво на производителност трябва да постигне устройството, за да се счита за валидирано?
- Планът за статистически анализ: как се обработват липсващите данни, как се изчисляват доверителните интервали, какви статистически тестове се използват
Минималният приемлив праг на производителност заслужава специално внимание. „Толкова добър, колкото предиката“ не е достатъчно, ако твърдите клинична полза. „Клинично не по-нисък от [бенчмарк]“ изисква дефиниране на границата на не по-ниска ефективност. Прагът на производителност трябва да бъде клинично значим – не само статистически значим – и обоснован от клинични доказателства за това какво ниво на производителност е адекватно за безопасна клинична употреба.
Ако вашето валидационно проучване е проведено без предварителна спецификация, това не дисквалифицира автоматично резултатите, но значително отслабва доказателствата и ще трябва да бъдете прозрачни относно това в доклада си за клинична оценка. По-важното е да планирате всички по-нататъшни валидационни проучвания с предварителна спецификация от самото начало.
Изисквания за набор от данни: какво всъщност търсят нотифицираните органи и FDA
Представителност на населението
Вашият набор от данни за валидиране трябва да включва пациенти, представителни за предвидената от вас популация. „Предназначена популация“ означава пациентите, към които вашият алгоритъм ще бъде приложен в клиничната практика — която може да е по-широка от популацията от пациенти в болницата, където сте събрали данните за обучение.
Ако вашият сърдечен изкуствен интелект е обучен и валидиран върху пациенти от голяма градска учебна болница в Западна Европа и възнамерявате да го внедрите в различни европейски клинични условия, вашият набор от данни за валидиране трябва да отразява това разнообразие – по възраст, пол, етническа принадлежност, профил на съпътстващи заболявания и условия на придобиване (тип скенер, качество на изображението, вариации в клиничния работен процес).
Нотифицираните органи все по-често очакват да видят статистически анализ на производителността в съответните подгрупи, а не само на обобщената производителност. Ако вашият алгоритъм е с чувствителност 94% при бели европейски пациенти и 81% при южноазиатски пациенти, това неравенство в производителността е регулаторен въпрос, а не просто въпрос на справедливост.
Независимост на данните за обучение и валидиране
Това е най-технически критичното изискване за данните и най-често нарушаваното на практика. Вашият набор от данни за валидиране трябва да бъде напълно независим от вашия набор от данни за обучение – не само на ниво случай, но и на ниво пациент. Ако пациент А има три сканирания във вашия набор от данни и едното сканиране е в процес на обучение, а две са във валидиране, данните на пациент А са попаднали във вашата валидация. Моделът може да е научил специфични за пациента модели, които се появяват като обобщение при валидирането, но не биха се обобщили за нови пациенти.
За ретроспективни проучвания върху болнични набори от данни, налагането на независимост на ниво пациент изисква изрично управление на идентификатора на пациента във вашия конвейер за данни. Ако не сте направили това стриктно, отчетената от вас ефективност на валидирането е оптимистична с неизвестна стойност.
Последователни или произволно подбрани случаи
Отклонението при селекцията във вашия набор от данни за валидиране е значителна заплаха за валидността. Избирането на „интересни“ или „ясни“ случаи води до оценки на производителността, които не отразяват съчетанието от случаи в реалния свят. За защитимо валидационно проучване е необходимо последователно вземане на проби от случаи (всички случаи, отговарящи на критериите за включване за определен период) или правилно рандомизирано вземане на проби.
Качество на истината
Производителността на вашия алгоритъм може да бъде измерена само спрямо фундаментална истина. Качеството на вашите валидационни доказателства е ограничено от качеството на вашите етикети за фундаментална истина.
За образната диагностика, основната истина обикновено е експертна рентгенологична анотация, но експертната анотация има вариабилност между оценители. Въпросът, който регулаторните органи задават: как се е обработвало несъгласието по анотацията? Ако даден случай е бил неясен, какъв е бил процесът на разглеждане? Имало ли е референтен стандарт (биопсия, резултат от проследяване, консенсус на комисията) за неясни случаи?
За предсказващите модели (прогнозиране на повторен прием, прогнозиране на началото на заболяването), основната информация идва от клиничните резултати в досиетата. Въпросът: резултатите ли са били записани последователно и пълно? Имало ли е различно проследяване между групите пациенти – някои пациенти са по-склонни да бъдат проследявани от други – което би могло да повлияе на данните за резултатите?
Методологията, основана на достоверни данни, трябва да бъде документирана във вашия протокол на изследването и докладвана във вашата клинична оценка. Ако е слаба, вашите оценки за ефективност са ненадеждни и нотифициран орган с клиничен опит ще забележи това.
Изискването за множество обекти
Нотифицираните органи за диагностика с изкуствен интелект от клас IIa все по-често очакват валидиране на множество места. Това означава доказателства за ефективност от поне два до три независими клинични места, а не само от мястото, където е разработен алгоритъмът.
Обосновката е ясна: моделите с изкуствен интелект, обучени върху данни от една клинична среда, често се представят значително по-зле в други среди поради различни производители на скенери, модели или протоколи за придобиване на данни; различни демографски данни за пациентите и състав на случаите; различни клинични работни процеси и практики за запис на данни; различни канали за предварителна обработка на изображения.
Валидирането на един обект е най-добрият сценарий за производителност на внедряването. Валидирането на множество обекти е по-реалистична оценка. Разликата между двете е реална и понякога голяма — публикуваната литература документира спад в производителността от 10–20 процентни пункта, когато диагностичните системи с изкуствен интелект се оценяват на външни обекти.
Валидирането на едно място е най-често срещаният предотвратим пропуск, който наблюдаваме при прегледите преди подаване на заявление. Той генерира повече запитвания към нотифицирани органи, отколкото всеки друг проблем с клиничната оценка – и запитванията обикновено са фатални за първоначалния график.
За стартиращи компании без данни от множество обекти към момента на първоначалното поставяне на маркировката „CE“: бъдете честни относно това ограничение в доклада си за клинична оценка, предложете PMCF проучване, което ще събира многоцентрови доказателства след пускане на пазара, и обсъдете това проактивно с вашия нотифициран орган по време на предварителното подаване. Пропускането на ограничението е по-лошо от признаването му.
Проучвания на читателите и клинична полезност
За инструменти с изкуствен интелект, предназначени да улеснят клиницистите, като например подпомагане на вземането на решения в радиологията, интерпретация на ЕКГ, вторичен четец в патологията, най-силното доказателство не е просто диагностичната точност. Това е клиничната полезност : дали инструментът с изкуствен интелект действително променя вземането на решения от клиницистите към по-добро?
Златният стандарт за дизайн на изследването тук е проучването на читателите : панел от клинични читатели (обикновено 8–15) прочитат фиксиран набор от случаи два пъти – веднъж без инструмента с изкуствен интелект, веднъж с него. Анализът на сдвоени сравнения измерва дали четенията, подпомогнати от изкуствен интелект, са по-точни, по-бързи или по-последователни от четенията без изкуствен интелект. Резултатът определя количествено действителната клинична полза, която вашият инструмент предоставя, когато се използва по предназначение.
Проучванията върху читателите са значителни начинания — от 6 до 12 месеца от протокола до окончателния доклад, разходи в диапазона от 100 000 до 400 000 евро в зависимост от възнаграждението на читателя и обема на случаите. Но те предоставят най-защитимите доказателства за клинична полза, а FDA все по-често ги изисква за инструменти с изкуствен интелект, предназначени да улеснят рентгенолозите или други клинични читатели. Нотифицираните органи ги очакват за диагностика с по-висока степен на точност, особено съгласно MDCG 2020-1.
Ако проучването на читателя е непрактично за първоначална маркировка „CE“, документирайте защо, планирайте такова като PMCF и бъдете прозрачни относно разликата.
Предназначена употреба спрямо действителна употреба: проблемът с несъответствието
Вашето клинично валидиране е специфично за предвидената употреба, т.е. специфичната популация пациенти, клиничната обстановка и контекста на употреба, документирани във вашата декларация за предназначение и вашите инструкции за употреба.
Какво се случва, когато клиницистите използват вашия алгоритъм извън предназначението му? Диагностичен инструмент, валидиран за скрининг на възрастни пациенти, се използва за педиатрични пациенти. Алгоритъм, валидиран върху изображения от един производител на скенери, се използва върху изображения от друг. Алгоритъм, обучен върху данни от вторична медицинска помощ, се внедрява в първичната медицинска помощ.
От регулаторна гледна точка, употребата извън предназначението е отговорност на потребителя или институцията, която го използва, а не на производителя — при условие че вашите инструкции за употреба ясно посочват предназначението и неговите ограничения, обучението ви е адекватно и наблюдението ви след пускане на пазара улавя сигнали за употреба извън одобрените показания.
От гледна точка на клиничната безопасност, това си е ваш проблем, защото ефективността на вашия алгоритъм извън валидирания му обхват е неизвестна - и ако той причини вреда на пациента, репутационните и търговските последици падат върху вас, независимо от регулаторното разпределение на отговорността.
Практическият отговор е двоен: проектирайте обхвата на предвидената употреба така, че да бъде възможно най-клинично реалистичен (не го ограничавайте изкуствено, за да улесните валидирането, ако клиничната употреба ще бъде по-широка) и изградете механизми за наблюдение след пускане на пазара, които откриват кога вашето устройство се използва извън предвидения му обхват.
Слой на Закона на ЕС за изкуствен интелект за валидиране
За програмите, обвързани с ЕС, Законът за изкуствения интелект е в допълнение към изискванията за клинична оценка на MDR и добавя специфични задължения, които се припокриват с - но не са идентични - с това, което нотифицираните органи са очаквали в миналото.
Най-важните допълнения за клинично валидиране:
Управление на данните за обучение — Законът за ИИ изисква документирани доказателства за качеството на данните, представителността и тестването за пристрастия в обучителния набор, а не само във валидационния набор. Това ефективно разширява „валидационните доказателства“ назад до начина, по който сте конструирали данните за обучение на първо място. Документирането на състава на набора от данни, методологията за етикетиране и смекчаването на пристрастията трябва да бъде защитимо при оценката на съответствието със Закона за ИИ.
Прозрачност за внедрителите — вашата клинична оценка трябва да предостави на внедрителите (болници, клинични отделения) достатъчно информация за производителността, за да работят с устройството безопасно. Това обикновено означава разбивка на производителността по подгрупи, анализ на калибрирането и документирани ограничения, всичко това в инструкциите за употреба, предназначени за внедрителя.
Разпоредби за човешки надзор — за системи с изкуствен интелект, при които резултатите се преглеждат от клиницисти, Законът за изкуствения интелект изисква документирани механизми, чрез които клиницистите могат да отменят, интерпретират или отказват резултатите от ИИ. Клиничната оценка трябва да разгледа как се запазва човешкият надзор в реална клинична употреба.
За програми, които вече са преминали през оценка на съответствието с MDR, съответствието със Закона за изкуствения интелект (AI Act) се интегрира сравнително гладко. За новите участници, планирайте документацията по Закона за изкуствен интелект заедно с клиничната оценка на MDR от първия ден — преоборудването е значително по-скъпо от паралелното изграждане и на двете.
PMCF: валидирането, което продължава след пускането в експлоатация
Клинично проследяване след пускане на пазара (PMCF) се изисква за изделия от клас IIa съгласно EU MDR — клиничната ви оценка се простира отвъд първоначалната маркировка CE и продължава през целия пазарен живот на изделието.
PMCF е регулаторният механизъм, който събира данни за производителността в реалния свят – производителност в различни клинични условия, дългосрочна надеждност, производителност върху подгрупи, недостатъчно представени в доказателствата преди пускането на пазара, и откриване на промяна в разпределението. По-специално за продукти с изкуствен интелект, PMCF взаимодейства с дизайна на PCCP (Предварително определен план за контрол на промените): данните, събрани чрез PMCF, могат да подпомогнат решенията за преквалификация, а актуализациите на моделите, активирани от PCCP, могат да бъдат валидирани чрез инфраструктурата на PMCF.
Надежден план за клинично изпитване след клиничен преглед (PMCF) в първоначалната ви клинична оценка подсилва представянето на информацията — той показва на нотифицирания орган, че разбирате валидирането като текущо задължение, а не като еднократно упражнение. Конкретни дейности по PMCF, които да се планират: проспективен мониторинг на ефективността на местата за внедряване, участие в регистъра, където е уместно, планирано събиране на доказателства за специфични подгрупи и изрични задействания за допълнителни клинични изпитвания, ако сигналите от мониторинга ги налагат.
За стартиращите компании практическото значение е, че инфраструктурата след пускане на пазара – наблюдение на производителността, обработка на жалби, докладване за бдителност, събиране на данни за PMCF – трябва да бъде проектирана преди одобрението, а не да бъде закрепена след това. Нотифицираните органи проверяват плана за PMCF по време на прегледа; един слаб план е червен флаг.
Как изглежда добрата документация за валидиране
Докладът за клинична оценка, който удовлетворява прегледа от нотифициран орган за диагностика от клас IIa с изкуствен интелект, обикновено включва:
- Декларация за предназначение — точна, пълна, прегледана от клиничен експерт
- Клинична информация — систематичен преглед на литературата за клиничното състояние, съществуващите диагностични методи и публикуваните данни за ефективността на изкуствения интелект
- Оценка на клиничните доказателства — за всеки клиничен елемент от данните, оценка на неговата релевантност, качество и принос към пакета от доказателства
- Първични данни за ефективността — вашето проспективно или ретроспективно валидационно проучване, с предварително зададени показатели, доверителни интервали, подгрупови анализи и сравнение с бенчмарк
- Най-съвременен анализ — как се представя вашето устройство в сравнение със съществуващите диагностични методи и сравними системи с изкуствен интелект
- Анализ на съотношението полза-риск — клинично заключение: доказателствата потвърждават ли, че ползата от използването на това устройство надвишава остатъчните рискове?
- План за PMCF — как ще продължите да събирате и анализирате клинични данни след пускането на пазара
Клиничният оценител, който подписва този документ, трябва да бъде квалифициран клиницист със съответния опит — или старши клиницист от вашия екип, или външен клиничен експерт, ангажиран за тази цел. Нотифицираният орган ще оцени квалификацията на клиничния оценител като част от прегледа.
Това, което отличава клинична оценка, която преминава, от такава, която не преминава, обикновено не е точността на модела, а дисциплината на предварителната спецификация, строгостта на популационния анализ, честността относно ограниченията и надеждността на плана за PMCF. Програмите, които използват ресурсите за клинично писане толкова внимателно, колкото и машинното обучение, се справят; програмите, които не го правят, не го правят.
Често задавани въпроси
There's no universal number. It depends on your condition prevalence, primary metric, and the confidence intervals you're claiming. For a binary classification task aiming to demonstrate sensitivity 90%+ and specificity 85%+ with reasonable 95% confidence intervals, you typically need at least 50 positive cases and 200 negative cases per condition. For rare conditions (STEMI, complete heart block), targeted enrichment to reach those numbers is required. Pre-specify the sample size with a statistical power calculation in your study protocol — notified bodies probe this.
Most Class IIa AI submissions are based on retrospective studies on prospectively collected data, which is acceptable when the prospective collection followed good clinical practice and the algorithm did not influence data collection. Plan a prospective PMCF study to run post-launch, document it in your clinical evaluation, and discuss the prospective gap proactively with your notified body during pre-submission. Omitting prospective evidence entirely is increasingly questioned by notified bodies for high-impact diagnostics.
The underlying expectations have converged. Both require pre-specified, independent, rigorously analysed evidence of clinical performance in the intended population. The differences are procedural and formatting: FDA is more prescriptive about study design for novel device types (especially De Novo), uses Pre-Submission interaction to refine evidence plans, and requires the evidence in FDA's specific format. EU MDR's CER follows MDCG 2020-1's structure. Clinical data can largely be shared between submissions; submission format is built independently.
For internal benchmarking, yes. For regulatory validation, generally no — public datasets are typically used for training or development, and using them for validation creates ambiguity about independence. Your validation dataset should be properly independent of training data and ideally collected specifically for validation. Public datasets can supplement as comparison points to published performance of analogous devices.
A reader study compares clinical reader performance with and without your AI tool. It's the gold standard for demonstrating clinical utility not just diagnostic accuracy, but actual impact on clinician decision-making. FDA increasingly requests reader studies for AI tools intended to augment radiologists or other clinicians. Notified bodies expect them for higher-acuity diagnostics. They're substantial undertakings, typically 12+ readers, paired-comparison analysis, 6–12 months of execution, but produce the strongest evidence of clinical benefit available.
Document the methodology and report the disagreement rates. Common mitigations include multi-rater consensus on training labels, separate adjudicated test sets where ground truth is clinically confirmed (downstream pathology, biopsy, outcome data), and panel review of ambiguous cases. Inter-rater kappa scores should be reported in the clinical evaluation. Treating ground truth as noisy and reporting it transparently is more defensible than pretending it isn't.
Notified bodies don't set absolute thresholds. Instead they assess whether your performance is clinically meaningful relative to existing standard of care and consistent with your intended use. For STEMI flagging, sensitivities above 90% with specificities above 85% are typical published targets. For atrial fibrillation detection in screening contexts, sensitivity 95%+ at specificity 95%+. Your clinical evaluation should justify the chosen targets against published benchmarks, not present them in a vacuum.
Partially. PMCF is required for Class IIa devices to continue gathering evidence post-launch and can fill specific gaps — real-world performance across diverse sites, long-term performance, rare event detection. But PMCF can't substitute for the initial validation evidence required for clearance. Your pre-submission evidence package needs to be defensible at submission; PMCF supplements it post-launch, it doesn't replace it.