



From Prototype to Class IIa: How to Get Your AI Diagnostic CE Marked Without Losing 2 Years

Eight months.
That’s how long it took KARDI AI to go from a working cardiac diagnostic prototype to EU Class IIa certification. Not two years. Not the 18+ months most founders are told to expect. Eight months with a team that had deep clinical expertise and a strong dataset, but no prior experience navigating EU MDR.
The difference wasn’t luck or a particularly accommodating Notified Body. It was a structured approach to the certification process that treated regulatory compliance as an engineering problem, not an administrative one.
This article is what we learned building that AI system, and what founders trying to do the same thing need to understand before they start.
Why most AI diagnostics stall in regulatory purgatory
The pattern is consistent across the MedTech startups we work with. A team builds something technically impressive like an algorithm that outperforms clinicians on a specific task, validated on a respectable dataset, with peer-reviewed publication interest. They raise a Series A, hire up, and start talking to NHS trusts or hospital systems. Then someone asks about CE marking.
At that point, one of three things happens:
They underestimate the scope. CE marking for a Class IIa AI diagnostic under EU MDR is not a checkbox exercise. It requires a clinical evaluation, a technical file running to hundreds of pages, a Quality Management System certified to ISO 13485, a risk management file compliant with ISO 14971, and software lifecycle documentation under IEC 62304. Most technical teams have built none of this. Most clinical teams don’t know it’s required.
They start regulatory work too late. “We’ll start the regulatory work in eight months when the product is more stable” is the most expensive sentence in MedTech. Regulatory requirements shape product architecture; they are not something you retrofit. Starting late means rebuilding things you’ve already built.
They hire the wrong help. Regulatory consultants who specialise in traditional medical devices often struggle with AI-specific requirements. The EU MDR contains specific provisions for software, but the practical interpretation, particularly around continuously learning algorithms, confidence outputs, and model versioning, is still being established through Notified Body precedent and MDCG working group documents. And as of 2026, you also need a partner who understands how the EU AI Act interacts with MDR for high-risk AI medical devices. You need someone who understands both the regulatory framework and the AI engineering.
The EU MDR classification question: where to start
The first decision is classification, and it’s more consequential than most founders realise.
Under EU MDR, AI diagnostic software is classified by its intended purpose and the severity of harm that could result from incorrect output. The relevant rule for most diagnostic AI is Rule 11 (software), which classifies software based on the decision it supports:
- Class IIa: Software intended to provide information used in decisions for diagnosis or therapy.
- Class IIb: Software providing information for decisions whose error could cause serious deterioration of health or surgical intervention.
- Class III: Software providing information for decisions that could cause death or irreversible deterioration of health.
Most diagnostic decision-support tools land at Class IIa or IIb. The distinction matters enormously. Under MDR, all Class IIa, IIb, and III devices require Notified Body involvement in the conformity assessment. There is no self-certification route as there was under the old MDD. What scales with classification is the depth of NB scrutiny: Class IIa typically follows Annex IX (full QMS audit plus technical file sampling) or the Annex XI Section 6 production assurance route; Class IIb requires NB review of technical files for representative devices in each generic group; Class III requires NB assessment of every individual technical file, plus the Annex IX scrutiny procedure with the relevant medical authority.
Getting the classification right before you design your regulatory strategy is not optional. We’ve seen teams spend eight months preparing a Class IIa technical file, only to have a Notified Body reclassify their device as IIb during initial review. The consequences are real: different evidence requirements, different audit scope, longer timelines, higher fees.
KARDI AI did the classification analysis first, before any regulatory documentation was started. Cardiac diagnostic support, where the algorithm flags ECG anomalies for cardiologist review, was confirmed as Class IIa under a structured Rule 11 analysis with the Notified Body’s pre-submission input. Knowing that with confidence shaped every subsequent decision.
The four pillars you need in place before touching the technical file
The technical file whch is the central regulatory dossier is where most teams start. It’s the wrong place. The file is the output of four systems that need to be built first.
1. Quality Management System (ISO 13485)
EU MDR requires that your QMS be certified to ISO 13485 before your device can receive a CE mark. This means an external certification audit by a Notified Body. The QMS covers how your organisation designs, develops, validates, and monitors software and it needs to be operational, not documented. Auditors will look for records of your processes, not just the procedures themselves.
The minimum viable QMS for a startup doing this for the first time is leaner than most consultants suggest. You don’t need the full enterprise framework a large medical device manufacturer runs. But you do need version-controlled procedures, design history files, change-control processes, and evidence that your team is following them. Building a QMS that looks right on paper but doesn’t reflect how you actually work is the single fastest way to fail a Notified Body audit.
2. Risk Management File (ISO 14971)
ISO 14971 requires a systematic risk management process across the full device lifecycle, from design through post-market. For AI diagnostics, this has specific implications that don’t apply to traditional software: how do you handle model uncertainty? What’s the failure mode when the algorithm encounters out-of-distribution data? How do you define and communicate acceptable risk thresholds for false negatives versus false positives in a clinical context?
The risk management file for KARDI AI required close collaboration between the clinical team (who understood the consequences of missed cardiac events) and the engineering team (who understood model behaviour at edge cases). That collaboration can’t be forced at the end of a project. It needs to be embedded in how the product is built.
3. Clinical Evaluation (MDR Annex XIV + MDCG 2020-1)
This is the evidence package that demonstrates your device performs as intended and is safe in its intended use. The primary references for MDR clinical evaluation are MDR Annex XIV and MDCG 2020-1 (clinical evaluation of medical device software); MEDDEV 2.7/1 Rev 4 remains a useful methodological reference but is no longer the binding guidance. For AI diagnostics, the clinical evaluation must include:
- A systematic literature review of the clinical background and existing diagnostics
- An appraisal of clinical data for your specific device, either from clinical investigations, post-market data, or equivalent-device equivalence arguments
- An analysis of clinical benefit versus residual risk
The equivalence route of claiming your device is equivalent to a predicate is significantly harder under EU MDR than under the previous MDD. For AI diagnostics, it’s rarely available, because the algorithm is typically novel. Most teams need actual clinical performance data, which means a prospective or retrospective clinical dataset with proper ground-truth labels, validated by qualified clinicians, with statistical analysis the Notified Body will accept.
For KARDI AI, the clinical dataset was the team’s strongest asset. The challenge was structuring the analysis to satisfy the specific evidence requirements, not generating the data. If you’re pre-data, this is where the timeline starts.
4. Software Lifecycle Documentation (IEC 62304)
IEC 62304 defines the software development lifecycle requirements for medical device software. For AI systems, this includes specific requirements around software architecture documentation, unit and integration testing, and critically for ML models, how you handle model training, validation, and updates.
The standard does not prescribe specific AI/ML methodologies, but your Notified Body will expect to see a documented approach to training/validation splits, hyperparameter selection, overfitting mitigation, and model versioning. MDCG guidance on software qualification and classification (MDCG 2019-11), on clinical evaluation of MDSW (MDCG 2020-1), and the ongoing MDCG working group activity on AI/ML provide the evolving regulatory context.
What about the EU AI Act?
Every AI MedTech founder is now asking: do I also need to comply with the EU AI Act? For a Class IIa diagnostic AI, the short answer is yes. The AI Act treats AI systems regulated as medical devices as high-risk, which adds a layer of obligations on top of MDR conformity:
- A documented AI risk management system that explicitly addresses AI-specific harms such as data quality, bias, drift
- Data governance requirements for training, validation, and test datasets
- Technical documentation aligned with AI Act Annex IV
- Logging and traceability of model outputs in production
- Transparency obligations toward users (clinicians, in this case)
- Human oversight provisions
- Robustness, accuracy, and cybersecurity requirements appropriate to the use case
The good news: a substantial portion of what you build for MDR such as risk management, technical documentation, software lifecycle, etc. also covers AI Act obligations. The bad news: not all of it. Data governance, bias monitoring, and transparency under the AI Act go further than what MDR alone requires, and the conformity assessment routes are designed to integrate with your Notified Body’s existing MDR scrutiny.
The pragmatic path is to build the MDR technical file first, but architect your data governance, model evaluation, and post-market monitoring infrastructure with AI Act obligations in mind from day one. Retrofitting AI Act compliance after CE mark is the same expensive mistake as retrofitting MDR compliance after product-market fit.
The Notified Body relationship: what nobody tells you
Notified Bodies are not adversaries. They are conformity assessment bodies that follow documented procedures — and if you understand those procedures, the review process becomes predictable.
The practical advice that makes the most difference:
Pre-submission meetings are worth more than they cost. Most Notified Bodies offer pre-submission consultations. For a novel AI diagnostic, this is not optional. You want to know how your specific Notified Body interprets Rule 11, what their expectations are for AI/ML clinical evaluation evidence, and whether they have reviewed similar devices before. A two-hour consultation will save you three months of rework.
Choose your Notified Body based on AI experience, not just cost. The Notified Body ecosystem varies significantly in its familiarity with AI medical devices. Some have dedicated AI review teams. Some are still building that capability. Check their published device portfolio. If they have never reviewed an AI diagnostic in your category, that is a risk to your timeline.
Submit a complete file, not a draft. Some teams submit partial files to “get feedback early.” Under EU MDR, this tends to backfire. Notified Bodies issue formal queries on submissions, and each query cycle takes time. A complete, well-structured submission, reviewed in advance, reduces the number of query cycles and compresses the timeline.
Post-market surveillance: the requirement that catches teams off-guard
CE marking is not the finish line. EU MDR has significantly strengthened post-market surveillance requirements, and for AI diagnostics, these are particularly demanding. The AI Act adds further post-market monitoring obligations specific to AI systems, including ongoing assessment of accuracy, robustness, and bias in deployment.
Your post-market surveillance plan must describe how you will:
- Collect and analyse field data from clinical use
- Monitor for unexpected performance degradation, including model drift in deployment
- Handle complaints and incidents
- Feed post-market data back into your clinical evaluation (PMCF: Post-Market Clinical Follow-Up)
- Manage model updates without triggering a new conformity assessment
The last point is where AI systems diverge from traditional software. If you update a conventional software feature, you assess the change under your QMS change-control process. If you retrain your AI model, even on new data without changing the architecture, the question of whether that constitutes a “significant change” requiring reassessment by your Notified Body is not fully resolved. MDCG guidance is evolving, and the safe approach is to have a documented change-assessment procedure that your Notified Body has reviewed and accepted in advance.
Building this infrastructure before you achieve CE mark, not after, is what allows you to iterate your model in a compliant way once you are on the market.
The timeline that’s actually achievable
For a Class IIa AI diagnostic starting from a working prototype with good clinical data:
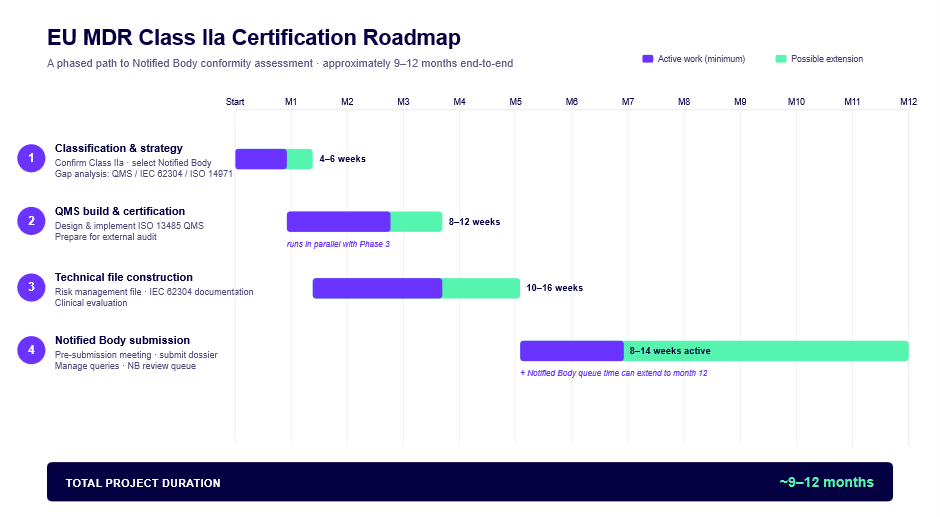
The eight months KARDI AI achieved sat at the fast end of this range, enabled by strong clinical data, a technically sophisticated team that could move quickly on documentation, and no significant reclassification surprises.
What makes timelines slip: discovering mid-process that your QMS doesn’t match how you actually work, clinical data that doesn’t satisfy the Notified Body’s evidence requirements, or a reclassification from IIa to IIb.
What to do before you start
Three questions to answer before any regulatory documentation begins:
What is your device’s intended purpose, precisely? Not the marketing version. The regulatory version: the specific clinical use case, the intended patient population, the intended user, the intended environment of use. EU MDR Article 2(12) defines this, and your classification, clinical evaluation strategy, and risk management all depend on getting it right.
Do you have the clinical data to support a Notified Body submission? If not, when will you, and is your dataset design appropriate for regulatory use? Random splits between training and validation sets are not acceptable if the same patient appears in both. Ground-truth labelling methodology will be scrutinised. Statistical power calculations matter.
Is your team structured to sustain a QMS? Not to build one for a certification audit and then ignore it but to actually operate it. The QMS requires designated responsibility, records, and ongoing compliance. If you don’t have a regulatory affairs lead, you need either to hire one or to retain someone who can perform that function.
If the answers are clear, the path through EU MDR is navigable. If they are not, those are the problems to solve before the regulatory work starts.