



The 6-month roadmap: from healthcare AI concept to regulatory submission


Most healthcare AI founders we speak to are not missing ambition, clinical knowledge, or technical capability. What they're missing is a clear sequence.
They know they need CE marking. They know they need clinical validation. They know they need a QMS. What they don't know is what to do first, what can run in parallel, what will block everything else if it's skipped, and what the realistic timeline looks like when you're not a 500-person medical device manufacturer with a dedicated regulatory affairs department.
This is that roadmap. It's based on what we've learned building regulatory-grade AI for healthcare companies — including taking KARDI AI's cardiac diagnostic from prototype to a complete EU Class IIa technical file submission in six months. Notified body review and certificate issuance happen after submission and add another 6–12 months on top, depending on the body's queue and your query cycles. This article is about the six months you control.
It's not theoretical. It's the sequence that works.
This is the operational companion to our pieces on SaMD classification, the engineering reality of cardiac AI, and the business case for cardiac AI. If you're not yet sure whether you're a medical device, start with the classification piece. If you've decided to build, this is the timeline.
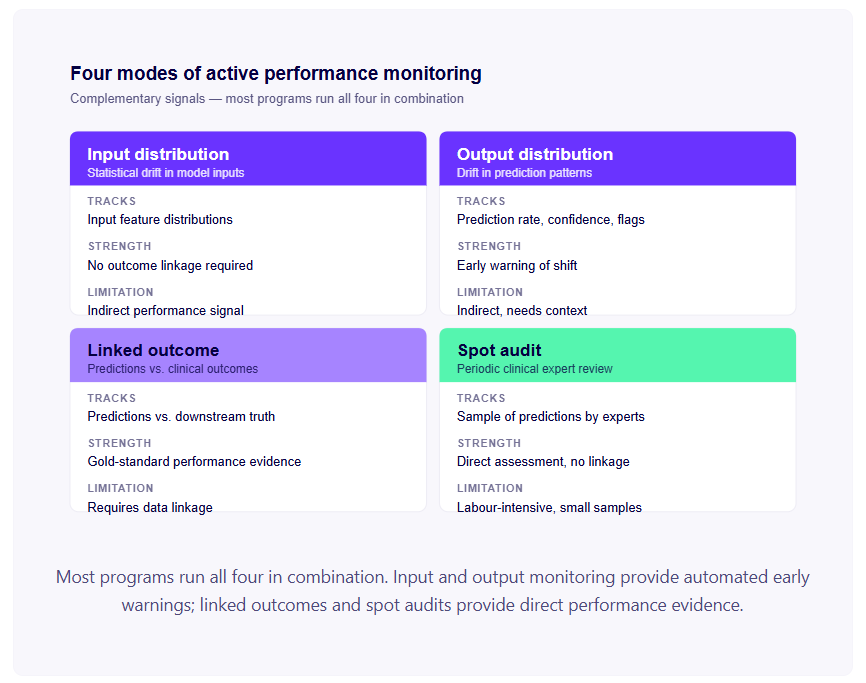
Before the clock starts: the prerequisites
Six months to submission is achievable. It's not achievable from zero. Before the roadmap begins, three things need to be true.
Your intended purpose is locked. The regulatory version, not the investor pitch version. Specifically: what clinical decision does your software inform? For whom? In what clinical setting? Used by whom? EU MDR Article 2(12) defines intended purpose, and every downstream regulatory decision — classification, clinical evaluation strategy, risk management scope — follows from it. Changing your intended purpose mid-process is expensive and frequently fatal to the timeline.
Your clinical dataset exists and is appropriate. Not just large — appropriate. Patient-level data with validated ground truth labels. No patient appearing in both training and validation sets. A labelling methodology a notified body will accept (multi-rater consensus where applicable, adjudicated test sets where outcome data exists). Statistical power sufficient to support the performance claims you'll make in your clinical evaluation. If this data doesn't exist, the clock starts when it does, not now. Building a regulatory-grade dataset is itself typically a 12–18 month workstream and is not in this roadmap.
Leadership is aligned on what CE marking actually requires. Founders sometimes discover mid-process that their board expected CE marking to cost €50K and take four months, when reality for a Class IIa is €300K–€800K of total program cost across 12–24 months from a serious start, plus notified body fees often €100K–€300K. That misalignment kills projects. Set expectations before the roadmap begins.
Month 1: classification, gap analysis, and strategy
Week 1–2: regulatory classification
Determine your EU MDR classification before anything else. For software, Rule 11 in Annex VIII is the relevant rule. Default classification is Class IIa. Class IIb applies if the consequence of the software-supported decision could be serious deterioration of health or surgical intervention. Class III applies if the consequence could be death or irreversible deterioration. Most clinician-in-the-loop AI diagnostics land in Class IIa.
Document your classification rationale in writing. This document becomes part of your technical file, and the argument needs to be defensible to a notified body who may disagree with you. Genuine borderline cases warrant a written classification opinion from a notified body, MDCG borderline guidance reference, or a competent authority consultation — formal mechanisms, not informal advice. (For a deeper treatment of classification logic across jurisdictions, see our SaMD classification piece.)
If you're targeting the US market simultaneously, map your device against FDA's software function guidance and determine whether you're 510(k) (if a substantially equivalent predicate exists) or De Novo (if not). PMA is rarely the right path for SaMD and you can usually rule it out quickly. Evidence requirements differ between EU and US, but the clinical data needed often overlaps.
Plan the AI Act overlay simultaneously. As of August 2026, EU MDR Class IIa and above devices that include AI systems are automatically classified as high-risk under the EU AI Act. The AI Act conformity assessment integrates with MDR but adds documentation around training data governance, transparency to deployers, human oversight provisions, and AI-specific post-market monitoring. Treat AI Act compliance as a parallel workstream from week one, not a retrofit.
Week 2–3: notified body selection
Not all notified bodies are equal for AI diagnostics. The EU MDR notified body ecosystem has roughly 50 designated bodies, but a much smaller number have meaningful experience reviewing AI/ML medical device software.
Request information from three to five notified bodies. Ask specifically: have they reviewed AI/ML software in your category (radiology, cardiology, diagnostics, etc.)? What is their current review timeline for a Class IIa technical file from submission to certificate? Do they offer pre-submission consultations? What are their specific expectations for AI/ML clinical evaluation evidence? Are they prepared for the AI Act conformity assessment integration?
Select on AI experience and capacity, not fee alone. Notified body capacity is currently a real bottleneck: some bodies have 12+ month waits just to begin a conformity assessment. Engage early; engaging at month five rather than month one regularly costs programs an entire quarter.
Week 3–4: gap analysis
Audit current state against six requirements:
QMS maturity. Documented procedures for design control, change management, document control, supplier management, complaints, and corrective action — and records showing you're running them, not just that you've written them.
IEC 62304 compliance. Software development lifecycle documentation. Architecture documentation. Unit, integration, and system test records. A classified software safety class for your system.
ISO 14971 risk management. Systematic identification and assessment of risks, including AI-specific failure modes (false positives, false negatives, distributional shift, calibration drift, alert fatigue).
IEC 62366-1 usability. Use specification, user profile, and a plan for formative and summative usability evaluations.
IEC 81001-5-1 cybersecurity. Threat modelling, secure development lifecycle documentation, vulnerability management, and security update distribution. This is increasingly a critical workstream and often underweighted by ML-led teams.
Clinical evidence and labelling. Dataset meeting the requirements above; pre-specified statistical analysis plan; an IFU that accurately describes intended purpose, intended users, contraindications, and limitations.
The gap analysis output is a prioritised work plan for months two through five. Don't skip it. Teams that skip it discover in month four that they have a six-month problem.
Month 2: QMS build and foundation
The Quality Management System is the foundation everything else sits on. If your QMS is non-compliant, nothing downstream matters.
What the QMS must cover, minimum, for a startup:
Document control — Version-controlled procedures and records. This doesn't require expensive software; it requires discipline and a system everyone follows.
Design and development — A documented design history file showing how your device evolved, with design inputs, outputs, verification, and validation records.
Software change control — A formal process for assessing and approving changes to your AI model, training pipeline, and software architecture. Especially important for ML systems where retraining constitutes a form of change. This is also where you decide whether to operate as a locked model or to plan for a Predetermined Change Control Plan (PCCP) — a strategic decision worth making early because it shapes the entire change-management story for the lifetime of the product. (FDA formalised PCCPs in 2024; EU regulators are following.)
Risk management integration — Your QMS must reference your ISO 14971 process and ensure post-market risks feed back into the risk file.
Supplier management — Cloud infrastructure, third-party model components, and data processing tools are suppliers under your QMS and require qualification.
Complaints and vigilance — A system for receiving, recording, and responding to field complaints and reportable incidents.
Cybersecurity processes — Vulnerability management, security incident response, and security update distribution. Increasingly a notified body focus area.
By end of month two, your QMS should be operational, not just documented. Notified body audits look for evidence you're running these processes, not just that you've written them down.
For a Class IIa MDR submission you don't strictly need separate ISO 13485 certification — your notified body will audit your QMS as part of conformity assessment. Many companies pursue separate ISO 13485 certification anyway to streamline the notified body audit, demonstrate maturity, and support US market access. If you're going that route, book your certification audit for month four. Most certification bodies need four to six weeks' notice.
Month 3: technical file — phase 1
Month three runs risk management, IEC 62304 documentation, and cybersecurity documentation in parallel with ongoing QMS operation.
Risk management file (ISO 14971)
For AI diagnostics, risk management goes beyond conventional software risk assessment. The hazards you need to address:
False negative rates and their clinical consequences. False positive rates and the cost of unnecessary follow-up. Performance on out-of-distribution inputs (populations or acquisition contexts not represented in training data). Model confidence calibration — does the algorithm's confidence score reflect its actual accuracy? Distributional shift over time as clinical practice evolves. Alert fatigue from over-flagging.
Mitigation measures: UI design that communicates uncertainty appropriately to clinicians; contraindications and population exclusions stated in the IFU; calibrated and condition-specific thresholds; post-market monitoring infrastructure to detect performance degradation; clear escalation pathways for low-confidence outputs.
The risk management file is a living document — it gets updated as you gather post-market data — but the initial version needs to be complete for submission.
IEC 62304 documentation
Software safety class follows from your risk classification. Class A if no injury is possible. Class B if non-serious injury is possible. Class C if death or serious injury is possible. Most diagnostic decision support tools land in Class B or C — Class C is more common than founders assume, because the consequence of an undetected serious condition (through clinician error or alert dismissal) qualifies.
IEC 62304 documentation for AI includes a software development plan; software architecture document covering ML model architecture, training pipeline, and inference pipeline; software unit, integration, and system test records; and an anomaly tracking system.
ML-specific documentation that notified bodies increasingly require: training/validation/test split methodology with patient-level isolation; dataset provenance, labelling methodology, and preprocessing steps; hyperparameter selection rationale; model versioning and change-assessment procedure; performance metrics broken down by clinically relevant subgroups (sex, age, comorbidities, acquisition equipment).
IEC 81001-5-1 cybersecurity
Cybersecurity documentation is a parallel workstream now, not an afterthought. The expected artefacts: threat model addressing the device's deployment environment; secure development lifecycle integrated into your QMS; vulnerability management process with documented scanning, triage, and remediation; security update distribution mechanism; security incident response plan tied to MDR vigilance reporting.
For networked AI medical devices — virtually all of them — cybersecurity findings are a leading source of notified body queries. Build the documentation alongside the technical file, not after.
Month 4: technical file phase 2 + usability + QMS audit
Clinical evaluation
The clinical evaluation is the evidence package demonstrating safe and intended performance. For a Class IIa AI diagnostic, it includes:
A systematic literature review covering clinical background, existing diagnostic methods, and published performance data for comparable AI systems. Documented methodology (typically PICO framework) with explicit inclusion/exclusion criteria. This is not a literature search.
Device-specific clinical data analysis demonstrating performance in your intended use population. Primary metrics (sensitivity, specificity, AUC, PPV, NPV) pre-specified, not chosen post-hoc. Subgroup analyses for clinically relevant variables (age, sex, comorbidities, acquisition equipment) increasingly expected. External validation evidence — performance on data not represented in training — substantially strengthens the file.
Clinical benefit vs. residual risk assessment — the conclusion regulators actually care about. Not "our algorithm performs well" but "the clinical benefit of deploying this device in clinical practice outweighs the residual risks given available mitigations."
Post-market clinical follow-up (PMCF) plan documenting how you'll continue gathering clinical evidence post-launch. PMCF is a regulatory requirement for Class IIa under MDR, not optional.
The clinical evaluation is signed by a clinical evaluator — a qualified clinician with relevant expertise. If that's not someone on your team, they need to be retained.
Usability evaluation (IEC 62366-1)
Usability is its own conformity assessment workstream. For AI medical software, the core questions: do clinicians correctly interpret the device's outputs? Do they understand its limitations and confidence levels? Do they know when to trust the output and when to override it?
Typical structure: formative evaluation during development (informal user testing to refine the interface), then summative evaluation closer to submission (structured testing with representative users in conditions approximating real use). Findings drive interface and IFU revisions, which feed back into the technical file.
For AI specifically, usability findings around uncertainty communication, alert fatigue, and clinician trust are increasingly probed by notified bodies. Treat this as a substantive evaluation, not a checkbox.
QMS audit
Whether you're going through ISO 13485 certification or directly into the notified body QMS audit, the audit typically runs two to three days. Common findings that delay completion: procedures exist but records show they're not being followed; change control records are incomplete for recent software updates; supplier qualification is underdocumented for key components (cloud infrastructure, third-party libraries, data labelling vendors); post-market surveillance plan is a generic template rather than specific to your device.
Address findings promptly — most auditors allow a 30-day window to respond to minor non-conformities before issuing the certificate or proceeding.
Month 5: pre-submission and notified body interaction
Pre-submission meeting
Before formal submission, book a pre-submission consultation with your notified body. Bring:
Your classification rationale document. A summary of your clinical evidence and analysis plan. Your draft clinical evaluation structure. Your AI Act conformity approach. Specific questions on AI/ML interpretation where notified body guidance is unclear.
The output is informal feedback you can use to refine your submission. It's not binding, but it significantly reduces the risk of unexpected queries post-submission. Pre-submission engagement done well saves 8–16 weeks of later query cycles.
Technical file review
Before submission, have your file reviewed by someone who has been through a notified body review before — ideally in your device category. Common issues that generate queries:
Intended purpose statement that's too broad or too narrow. Clinical evaluation that doesn't address the notified body's specific evidence questions. Risk management that doesn't adequately address AI-specific failure modes. IEC 62304 documentation that describes the process but doesn't provide evidence it was followed. Cybersecurity documentation that lists procedures without evidence of execution. AI Act documentation that's been bolted on rather than integrated. Clinical evaluation lacking external validation or subgroup analysis.
Every query cycle adds four to eight weeks to your post-submission timeline. The investment in a pre-submission review pays back in months saved.
Data protection alignment
Confirm your GDPR documentation (Data Protection Impact Assessment, lawful basis, data processing agreements, security controls) is consistent with the technical file claims about training data governance and post-market data flows. A GDPR–MDR documentation mismatch is a needless source of audit findings and can stall the program independently of medical device review.
Month 6: formal submission and the path beyond
Submission
A Class IIa technical file submission is a structured document package — typically several hundred pages plus annexes covering QMS records, clinical evaluation, risk management, IEC 62304, cybersecurity, usability, AI Act conformity, and labelling. Submit complete. Do not submit a draft and rely on query cycles to fix gaps; that strategy roughly doubles the time-to-certificate.
Query management
Most notified bodies operate on a formal query/response cycle. Your initial submission is reviewed (typically within 6–10 weeks for a body with capacity), queries are issued, and you respond. For a well-prepared submission to a notified body with AI experience, one to two query cycles is normal. Three or more indicates a significant gap in the submission and is the leading reason programs miss their first revenue year.
Treat queries as engineering problems. Read each query carefully, identify exactly what evidence or clarification is being requested, and respond with precision. Generic responses that don't directly address the query extend the cycle.
For AI-specific queries — increasingly common as notified bodies build expertise — the relevant guidance corpus includes MDCG 2019-11 (qualification and classification of software), MDCG 2020-1 (clinical evaluation/performance evaluation of medical device software), MDCG 2019-16 (cybersecurity), Team-NB position papers on AI/ML, and AI Office guidance on the AI Act. Your reviewer is working from this corpus; you should be too.
After submission
Submission is the start of a new clock, not the end. Realistic expectations from submission to certificate for a well-prepared Class IIa AI submission: 6–12 months including query cycles, depending on notified body capacity and the complexity of any AI-specific evidentiary disputes. Pessimistic scenarios with capacity-constrained bodies and multiple query cycles can run 12–18 months.
Plan commercial activities — clinical partnerships, hospital pilots under investigational arrangements, US submission running in parallel — to use this window productively rather than waiting for the certificate.
What the six-month target actually requires
Achieving six months from concept to a complete, well-prepared submission requires:
Starting with a complete, validated, regulatory-grade clinical dataset. The roadmap above doesn't include data acquisition because data acquisition takes 12–18 months on its own.
Running QMS build, risk management, IEC 62304 documentation, cybersecurity, usability, and AI Act conformity in parallel from months two through four — not sequentially.
A designated regulatory lead (internal or retained) who owns the process full-time. Half-time regulatory leads produce half-finished submissions.
Getting classification right in month one so nothing has to be reworked.
Treating the technical file as the product. In regulatory terms, it is.
Submitting a complete, reviewed file rather than an iterative draft.
None of this is out of reach for a Series A or B company. But it requires treating regulatory compliance as a product function, not an administrative afterthought — and starting early enough that the timeline doesn't become a crisis.
Where Vector Labs fits
We work with healthcare AI founders and med tech companies on regulatory program execution at three points: program scoping (sequencing, timeline reality, build/buy decisions on regulatory infrastructure), technical and clinical documentation (technical file construction, clinical evaluation, AI Act conformity), and notified body interaction (pre-submission, query response, audit preparation).
If you're scoping a regulatory program — internally or as part of a build/buy decision — and want to pressure-test the timeline against what we learned shipping KARDI AI, get in touch at vector-labs.ai.
For the broader context, the four-article set: SaMD classification covers what to determine before you start this roadmap; the engineering reality of cardiac AI covers what clinical-grade actually requires; the business case for cardiac AI covers the strategic frame around the regulatory work.