



Post-Market Surveillance for AI Medical Devices: Why CE Marking Is the Start, Not the Finish


CE marking is a milestone. It is not a finish line.
For most traditional medical devices, post-market surveillance is a systematic but relatively passive activity — collecting complaint data, monitoring adverse events, reviewing literature, and updating the clinical evaluation periodically. For AI medical devices, post-market surveillance is a fundamentally active and continuous programme — because the relationship between your device and the real world changes over time in ways that traditional devices don't experience.
The data distribution that clinical practice generates shifts. Patient populations change. Imaging equipment is upgraded. Clinical workflows evolve. The model that was validated in 2024 on data from three clinical sites may perform materially differently in 2026 on data from twenty clinical sites serving different patient populations with different equipment.
This is not a hypothetical risk. Published research documents performance drops of 10–20 percentage points when AI diagnostic systems are evaluated on external datasets not represented in training. Extrapolating to real-world deployment diversity and multi-year time horizons, the risks are real and require active management.
This is the post-market companion to our pieces on clinical validation (the pre-market evidence base that post-market surveillance extends), how to write a technical file (where the PMS plan lives in Annex III), the six-month regulatory roadmap (where PMS infrastructure should be built), and the engineering reality of clinical-grade AI (the technical monitoring infrastructure behind active surveillance).
What EU MDR Requires for Post-Market Surveillance
EU MDR Annex III defines the post-market surveillance requirements. The core obligations:
A Post-Market Surveillance Plan. Written before CE marking, describing how the manufacturer will proactively collect and analyse data on the performance and safety of the device throughout its commercial lifetime. For AI devices, the plan must specifically address how ongoing performance of the AI/ML system will be monitored.
A Post-Market Surveillance Report (for Class IIa). An annual summary of the post-market surveillance data, conclusions, and any actions taken. The report includes: summary of complaints and adverse events, results of any post-market clinical follow-up, assessment of the benefit-risk balance, and any conclusions about the need for updates to the clinical evaluation or risk management file.
A Periodic Safety Update Report (PSUR). For Class IIa devices, a PSUR is submitted to the notified body at minimum every two years. For Class IIb and III, at least annually. The PSUR summarises the post-market surveillance results and the conclusions for the benefit-risk profile of the device.
Post-Market Clinical Follow-Up (PMCF). Where the initial clinical evaluation has limitations — limited sample size, single-site validation, short follow-up period — the manufacturer must conduct PMCF to fill those gaps post-market. For AI devices submitting with limited multi-site data, a PMCF plan proposing real-world performance studies is typically expected.
Vigilance reporting. Serious incidents (a malfunction or deterioration in characteristics leading to patient death or serious deterioration of health) must be reported to the relevant competent authority within defined timelines under MDR Article 87 — 15 days for serious incidents, 10 days for death or unanticipated serious deterioration, and 2 days for serious public health threats. For AI devices, a serious incident includes patient harm directly attributable to a false negative or false positive from the algorithm.
EUDAMED registration. Devices and manufacturers must be registered in EUDAMED, and post-market data flows (PSUR submission, serious incident reports, field safety corrective actions) feed into the database. The PMS infrastructure needs to align with EUDAMED submission formats and timelines.
The AI-Specific Post-Market Surveillance Problem: Distributional Shift
Traditional medical device performance is stable unless the physical device degrades or is modified. An ECG monitor that works correctly today will work correctly next year, assuming it's maintained and calibrated.
AI model performance is not stable in this way. The model produces outputs based on the statistical patterns learned from training data. If the distribution of inputs the model receives in production shifts away from its training distribution, performance can degrade without any change to the model itself.
Distributional shift in medical AI occurs from:
Scanner and equipment changes. Hospitals upgrade imaging equipment on cycles of 5–10 years. A mammography AI validated on images from Scanner Model X may perform differently on images from Scanner Model Y, even if both produce diagnostically equivalent images to a radiologist. The model learned texture and contrast patterns specific to the training scanner.
Clinical workflow changes. If the clinical protocol for ordering an ECG changes — shifting from selective ordering to broader screening — the case mix arriving at the AI system changes. A model validated on symptomatic patients may receive a higher proportion of asymptomatic patients with lower disease prevalence, changing its positive predictive value substantially.
Population changes. Patient demographics change over time — aging populations, migration, changing disease patterns. A model trained on a 2022 patient population may encounter systematically different input distributions by 2026.
Seasonal and temporal variation. Some conditions have seasonal patterns (influenza, certain respiratory presentations). A model trained on data from one time period may see different input distributions at different times of year.
Annotation practice changes. If the labelling conventions for the ground truth in your training data reflect 2023 clinical practice guidelines, and those guidelines are updated in 2025, the model's predictions may systematically diverge from the new standard — not because the model changed but because the definition of ground truth changed.
What Active Performance Monitoring Looks Like
For an AI medical device, "monitoring complaints" is not a sufficient post-market surveillance strategy. You need active performance monitoring that detects distributional shift and performance degradation before it produces patient harm.
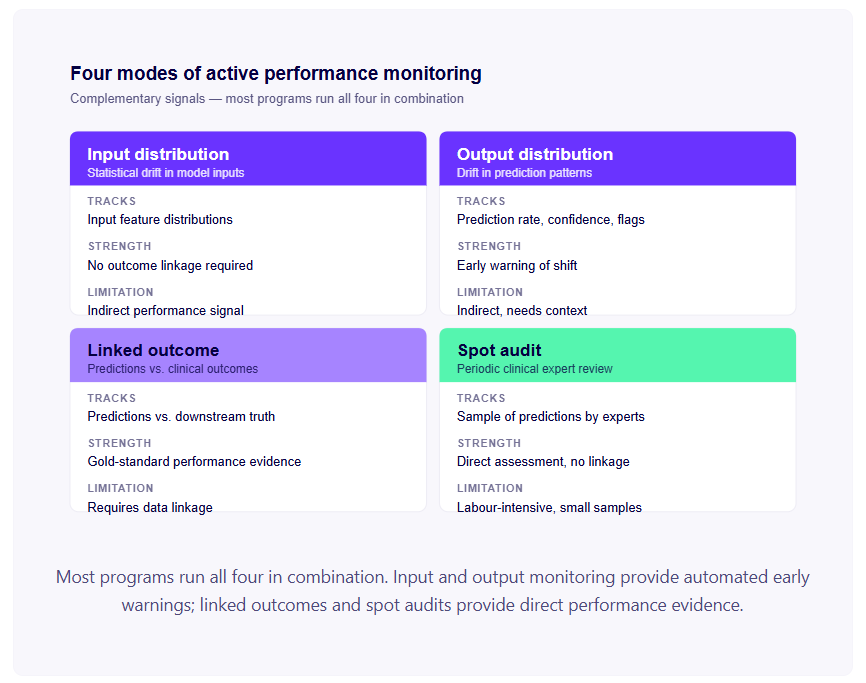
Input distribution monitoring. Statistical tracking of the distribution of inputs the model receives in production. For imaging AI: track distributions of image quality metrics, acquisition parameters, patient demographics. For ECG AI: track distributions of heart rate, signal quality metrics, acquisition equipment identifiers. Statistical tests (population stability index, KL divergence, Kolmogorov-Smirnov tests) can detect when the input distribution has shifted significantly from the training distribution.
Input distribution monitoring doesn't require clinical outcome data — it's monitoring the data going into the model, not the correctness of its outputs. This makes it implementable without complex clinical data linkage, and it provides an early warning signal.
Output distribution monitoring. Tracking the distribution of model outputs — the proportion of cases flagged as high-risk, the distribution of confidence scores, the proportion of cases flagged as out-of-distribution. Changes in output distributions may indicate distributional shift in inputs, changes in clinical practice, or model performance degradation.
Linked outcome monitoring. Where clinical outcome data can be linked to model predictions — through EHR integration, clinical audit programmes, or formal PMCF studies — tracking the relationship between model predictions and subsequent clinical outcomes provides direct performance evidence. This is the gold standard for real-world performance monitoring, but it requires data linkage infrastructure and appropriate governance for accessing outcome data.
Spot audit. A periodic clinical review of a random sample of model predictions, with clinical expert assessment of whether the predictions were correct. Labour-intensive but provides direct performance evidence without requiring automated data linkage.
Post-Market Clinical Follow-Up (PMCF): Filling the Evidence Gaps
PMCF is the planned, systematic collection of clinical data post-market to fill gaps in the evidence base from the initial clinical evaluation. For AI medical devices, PMCF is typically required where:
- The initial validation was single-site and multi-site performance data is needed
- The validation dataset had limited representation of specific demographic groups
- The follow-up period in the validation study was insufficient to assess long-term performance
- The clinical environment in which the device is deployed differs from the validation environment
The PMCF plan (submitted as part of the CE mark technical file) describes the planned studies, their objectives, their methodology, and the timeline for completion. The PMCF evaluation report (updated periodically, typically annually) summarises the results and their implications for the device's benefit-risk profile.
For AI devices, common PMCF designs:
Multi-site performance study. Deploying the device at additional clinical sites not included in the initial validation and collecting prospective performance data. The primary objective is usually to confirm that performance in the validation sites generalises to a broader range of clinical environments.
Long-term cohort follow-up. Following patients whose cases were assessed by the AI system and tracking clinical outcomes over an extended period. Particularly relevant for diagnostic AI where the clinical consequence of false negatives may only become apparent months or years later.
Specific subgroup studies. Targeted data collection for demographic groups underrepresented in the initial validation — specific age groups, ethnic backgrounds, clinical comorbidities.
Model Update Management: The Hardest Post-Market Problem
Every AI medical device manufacturer eventually faces the question: we want to improve our model. What does that require under EU MDR?
The EU MDR framework was not designed for iterative ML model improvement. Change-management requirements for new devices come from MDR Annex II/III (technical documentation maintenance), Article 10 (manufacturer obligations), and Article 56 (notified body re-assessment), plus ISO 13485 and IEC 62304 software change control. Together, these require assessment of whether a change to a CE-marked device constitutes a "significant change" requiring a new conformity assessment.
What constitutes a significant change for an AI medical device? EU MDR does not provide a clear answer. MDCG 2020-3 — originally guidance on significant changes under Article 120 transitional provisions for legacy MDD-certified devices — provides definitions that have been broadly adopted as the de facto framework. For AI devices, the question is being resolved gradually through notified body practice and emerging guidance, but as of 2026 there is no clear consensus.
The practical approaches:
A documented change assessment procedure. Define, in advance, criteria for classifying model updates as minor (within-scope performance improvement, no change to intended use, validated performance maintained), moderate (change to training data scope, change to model architecture, new clinical conditions added to scope), or major (change to intended use, substantial architecture change, significant performance change). Get your notified body to review and accept this procedure as part of your initial technical file.
The Predetermined Change Control Plan (PCCP) approach. Formalised by FDA in December 2024 for US submissions, increasingly referenced in EU regulatory discussions. Pre-specify the types of model updates you anticipate, the validation methodology you'll apply, and the performance criteria that confirm the update is safe. Obtain notified body acceptance of the PCCP as part of your initial submission. Then implement updates within the pre-approved scope without a new full conformity assessment.
Conservative approach for major updates. For changes outside your pre-defined scope — new intended use, new clinical indications, fundamental architecture changes — plan for a significant change assessment, which may require notified body review and may trigger a new conformity assessment. These are major product development milestones, not routine model maintenance.
The EU AI Act Post-Market Overlay
For EU-bound AI medical devices, the AI Act adds post-market obligations on top of MDR's PMS framework. As of August 2026, with high-risk AI Act provisions in force, these requirements apply to any Class IIa+ medical device that includes an AI system.
The relevant AI Act post-market obligations:
Post-market monitoring system (Article 72). A documented system for collecting, analysing, and acting on data about the AI system's performance throughout its lifecycle. The scope overlaps with MDR PMS but is framed specifically around AI system characteristics — bias, performance across subgroups, transparency to deployers, human oversight effectiveness.
Serious incident reporting (Article 73). The AI Act has its own reporting requirements for serious incidents related to AI systems, with timelines and recipient authorities that may differ from MDR vigilance reporting. For Class IIa AI medical devices, both regimes apply in parallel.
Substantial modification triggers (Article 43). The AI Act defines substantial modifications that trigger fresh conformity assessment for the AI component — overlapping with but not identical to MDR significant change criteria.
Lifecycle accuracy, robustness, and bias monitoring (Article 15). Ongoing documentation that the AI system continues to meet the accuracy, robustness, and bias thresholds it was assessed against at conformity assessment.
The practical implication: programs already operating under MDR PMS can integrate AI Act post-market obligations incrementally. New programs should plan both regimes' post-market documentation in parallel — separate reporting workflows but largely shared underlying data infrastructure.
Cybersecurity Post-Market Monitoring
Beyond MDR vigilance, networked AI medical devices face ongoing cybersecurity obligations under IEC 81001-5-1 and MDCG 2019-16 guidance. The post-market cybersecurity workstream includes:
- Vulnerability monitoring (CVE tracking, security advisories for dependencies)
- Security incident response (containment, communication, regulatory reporting where applicable)
- Patch management and distribution aligned to your software change control process
- Periodic threat re-assessment as the deployment environment evolves
For cloud-hosted AI medical devices, cybersecurity post-market monitoring is continuous — the threat surface changes constantly. Plan a separate cybersecurity surveillance workstream alongside the clinical PMS workstream.
The Post-Market Surveillance Infrastructure You Need Before CE Mark
Building post-market surveillance infrastructure after CE mark, under time pressure from the first compliance deadline, is significantly harder than building it as part of the device architecture. The minimum infrastructure that should be in place at CE mark:
Structured logging. Every model inference logged with: timestamp, input data identifier (anonymised), model version used, output (prediction and confidence), and any quality flags raised. Stored in a retrievable format accessible to your post-market surveillance team.
Complaint intake process. A formal mechanism for receiving, recording, and tracking complaints from clinical users, with defined response timelines and escalation criteria for potential serious incidents.
Vigilance procedure. A documented process for assessing potential serious incidents, determining reportability under EU MDR Article 87 and EU AI Act Article 73, and making timely reports to the relevant competent authorities.
PMCF data collection mechanism. If your PMCF plan involves prospective outcome data collection, the data collection mechanism — patient consent workflow, data linkage approach, storage, and governance — should be operational at the point of CE mark, not designed after.
Performance monitoring dashboards. Input and output distribution monitoring, anomaly detection, and threshold-triggered alerts should be operational from day one of clinical deployment. Retrofitting monitoring after performance has already drifted is too late.
Post-market surveillance is the workstream that most cardiac AI and medical AI programs build last and pay for first. Resourcing it as a parallel workstream before CE mark — not after — is the single highest-leverage post-market decision.
Where Vector Labs Fits
We work with AI medical device manufacturers on post-market surveillance at three points: pre-market PMS infrastructure design (logging, monitoring dashboards, complaint and vigilance workflows, PMCF data collection); ongoing PMS operation (PMS Report and PSUR construction, distributional shift detection, change-management decisions); and AI Act / MDR post-market integration (Article 72 monitoring systems, Article 73 incident reporting, AI Act conformity maintenance).
If you're scoping the PMS workstream for an AI medical device — or running into model-update questions post-launch — get in touch at vector-labs.ai.
For the broader series: SaMD classification covers whether you need a technical file at all; the six-month regulatory roadmap covers where PMS infrastructure should be built into the program; clinical validation covers the pre-market evidence base; how to write a technical file covers the Annex II and Annex III documentation structure; the engineering reality of clinical-grade AI covers the technical monitoring infrastructure.
FAQs
The PMS Report is an internal summary of post-market surveillance data, conclusions, and actions — required to be available for inspection but not routinely submitted. The PSUR (Periodic Safety Update Report) is a more comprehensive document submitted to your notified body, summarising post-market surveillance results and the device's benefit-risk profile. For Class IIa devices, the PSUR is required at least every two years; for Class IIb and III, at least annually.
Significant changes — those affecting the device's safety, performance, intended use, or essential design — typically require notified body involvement. For AI/ML, significant changes generally include changes to intended use, expansion to new clinical indications, fundamental model architecture changes, or material performance change. Within-scope retraining on new data, threshold adjustments, and minor performance improvements typically don't require a new conformity assessment if they're covered by a Predetermined Change Control Plan envelope accepted by your notified body.
Three common approaches. First, EHR integration where deployment sites share de-identified outcome data with the manufacturer under appropriate data-sharing agreements. Second, formal PMCF studies that prospectively collect outcomes for a sample of cases. Third, clinical audit programmes where clinical partners conduct periodic chart review and share aggregated performance data. Each requires upfront contracting and governance work that should be planned before CE mark.
At minimum: structured logging of every model inference (timestamp, input identifier, model version, output, quality flags); a complaint intake process with defined response timelines; a vigilance procedure documenting how serious incidents are assessed and reported; and the PMCF data collection mechanism if your plan includes prospective outcome collection. Retrofitting these after CE mark, under time pressure from the first reporting deadline, is significantly harder than building them as part of the device architecture.
The AI Act adds requirements on top of MDR for high-risk AI systems (which include MDR Class IIa+ medical device AI). Article 72 requires a documented post-market monitoring system specifically for the AI; Article 73 requires reporting of serious incidents within timelines defined by the Act; Article 43 defines substantial modifications that trigger fresh conformity assessment for the AI component. The AI Act and MDR post-market frameworks overlap but require separate documentation and reporting workflows.
FDA formalised PCCPs in December 2024. The EU regulatory framework is converging toward similar principles but doesn't yet have a formal PCCP equivalent under MDR. The practical approach: design your PCCP to satisfy FDA, document the same change-management plan in your MDR technical file (within IEC 62304 software change control and the PMS plan), and have your notified body review and accept the scope. The result is functionally equivalent in both jurisdictions.
Under EU MDR Article 2(65), a serious incident is any malfunction or deterioration in the characteristics or performance of a device that has led, directly or indirectly, to death, serious deterioration in health, or serious public health threat. For AI medical devices this includes patient harm directly attributable to a false negative (missed diagnosis leading to harm), false positive (unnecessary intervention with adverse consequences), or systematic performance degradation affecting multiple patients. Reporting timelines under Article 87: 15 days for general serious incidents, 10 days for death or unanticipated serious deterioration, 2 days for serious public health threats.
The plan is maintained continuously as a living document. The PMS Report is generated annually for Class IIa devices. The PSUR is updated at minimum every two years for Class IIa, annually for Class IIb and III. Significant changes in regulation, in real-world performance data, or in the device itself trigger updates outside the scheduled cadence.