



FDA 510(k) vs De Novo vs PMA: Choosing the right regulatory pathway for your AI medical device


FDA has cleared roughly 1,000 AI/ML-enabled medical devices through 2024, with more than 200 cleared in 2023 alone (per FDA's published list of AI/ML-enabled medical devices). Behind each of those clearances is a regulatory strategy decision that determined how long the process took, how much it cost, and whether it succeeded at all.
That decision is pathway selection: 510(k), De Novo, or PMA. For founders and CTOs building AI diagnostics targeting the US market, getting this choice right is the single most consequential early decision in your regulatory programme. Getting it wrong costs 12–18 months and often requires starting over.
This article explains the three pathways, what determines which one applies to your device, and how to build a strategy around the right one — including what EU MDR experience does and doesn't transfer.
This article assumes you've already determined your software is an FDA-regulated medical device. If you haven't, start with our SaMD classification piece — for many AI products (particularly non-image, non-signal CDS), the right answer is "no FDA submission required" and pathway selection is moot.
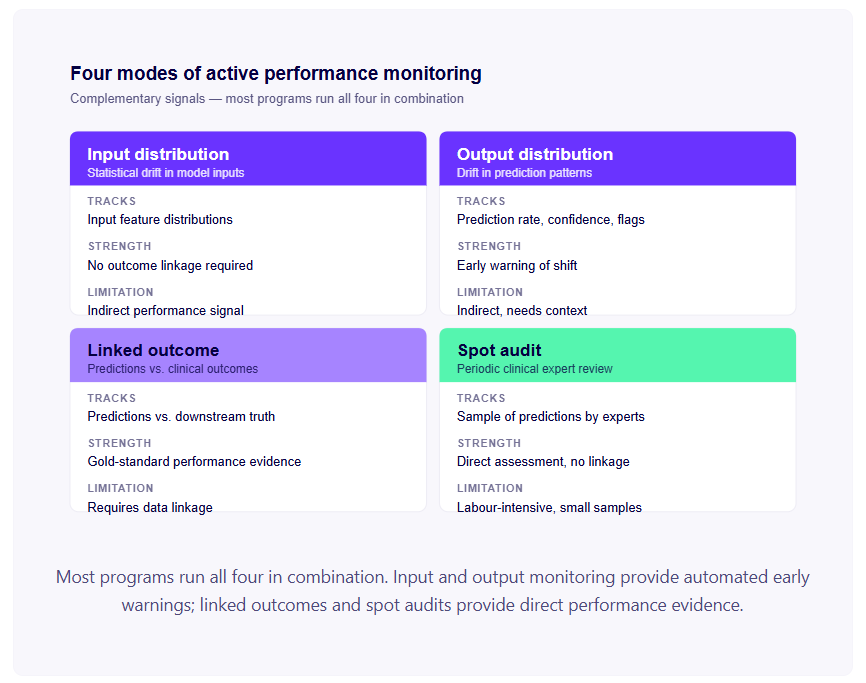
The framework: how FDA thinks about medical device risk
FDA classifies medical devices into three classes based on risk:
Class I — Low risk. General controls (basic regulatory requirements) are sufficient. Most Class I devices are exempt from premarket review. Examples: bandages, tongue depressors, most non-diagnostic software with no clinical decision support function.
Class II — Moderate risk. General controls plus special controls (performance standards, post-market surveillance, device-specific guidance). Most AI diagnostic decision-support tools land here. Requires either 510(k) clearance or, for novel device types without predicates, De Novo authorisation.
Class III — High risk. Involves supporting or sustaining life, or presenting a substantial risk of illness or injury. Requires Premarket Approval (PMA) — the most rigorous pathway. Examples: implantable cardiac defibrillators, certain automated diagnosis systems that replace clinical judgement.
The pathway determination starts with classification. Classification determines the regulatory standard. The regulatory standard determines what evidence you need.
A quick orientation before the detail: most AI diagnostic decision-support tools land in Class II. If a comparable AI device has already been cleared in your clinical category, your pathway is 510(k). If yours is the first AI device of its kind, your pathway is De Novo. PMA applies only to high-risk devices that directly drive treatment decisions or replace clinical judgement — rarely the right pathway for an AI startup.
Pathway 1: 510(k) — substantial equivalence
The 510(k) pathway is available when your device is substantially equivalent to a legally marketed predicate device. Predicates can be devices on the market before the Medical Device Amendments of 1976, devices reclassified from Class III to Class II, or devices that were themselves cleared through 510(k).
What substantial equivalence means: your device has the same intended use as the predicate, and either the same technological characteristics, or different technological characteristics that don't raise new safety and effectiveness questions and perform at least as well as the predicate.
For AI medical devices, the predicate question is the critical one. The AI device landscape has changed so rapidly that predicates now exist across most major clinical categories — radiology, cardiology, ophthalmology, pathology, dermatology. If you can identify a cleared AI device in your category with a comparable intended use, 510(k) is likely your pathway.
The evidence package for 510(k) AI devices
A typical 510(k) submission for an AI/ML device includes a 510(k) summary or statement identifying the predicate and the substantial equivalence argument; performance testing demonstrating your device meets or exceeds the predicate's performance; software documentation per IEC 62304 and FDA's software function guidance; AI/ML-specific documentation aligned with FDA's Good Machine Learning Practice (GMLP) principles, jointly published by FDA, Health Canada, and MHRA in October 2021; transparency documentation per the September 2022 CDS guidance and the December 2024 PCCP final guidance; clinical data (retrospective or prospective) demonstrating performance in the intended use population; and cybersecurity documentation per FDA's September 2023 cybersecurity guidance.
Three flavours of 510(k)
FDA offers three types of 510(k). Traditional 510(k) is the standard submission for a new device with a predicate. Abbreviated 510(k) applies when an FDA-recognised consensus standard or special control fully addresses the safety and effectiveness questions; the submission is lighter. Special 510(k) is the most relevant for AI/ML manufacturers: it covers modifications to your own previously cleared device where the change is well-characterised and doesn't affect the indications for use. For AI products making model updates or feature additions that fall outside a PCCP envelope, Special 510(k) is often the right vehicle — faster and lighter than a full new submission.
Timeline and cost
FDA has a 90-day statutory review target for 510(k)s, but the review clock pauses during additional-information requests. In practice, AI/ML device 510(k)s typically run 120–270 days from submission to decision, with the longer end reflecting multiple AI request cycles. Budget 6–9 months from submission and earlier for the preparation work.
FY2026 FDA user fees are approximately $25,000 standard or $6,000 small business. Total program cost, including consulting, clinical data work, and submission preparation, typically runs €300K–€1M+ depending on clinical data needs.
The predicate trap
The most common 510(k) failure mode for AI devices is a predicate argument that doesn't hold. Choosing a predicate with a similar-sounding name but a different intended use, or claiming substantial equivalence for a device that uses fundamentally different technology without adequate performance comparison, generates an Additional Information request or a Not Substantially Equivalent (NSE) determination. NSE is not a refusal to approve your device — it's a determination that your device can't use the 510(k) pathway, which routes you to De Novo. The cost is time, not the programme.
Pathway 2: De Novo — novel low-to-moderate risk devices
De Novo is the pathway for novel, low-to-moderate-risk devices that don't have a legally marketed predicate. It was designed specifically for the situation where a device type is genuinely new — there's nothing to be substantially equivalent to — but the risk level doesn't justify the full PMA burden.
When De Novo applies for AI devices:
You're building in a clinical category where no AI device has been cleared before. Or your AI approach is sufficiently different from cleared predicates that a substantial equivalence argument would not hold. Or your device is Class II risk despite its novelty.
The De Novo process
You submit a De Novo request demonstrating that your device is low-to-moderate risk and proposing the special controls that would provide reasonable assurance of safety and effectiveness. FDA reviews the request and, if granted, simultaneously authorises your device and creates a new device classification — which other manufacturers can then use as a predicate for future 510(k)s.
The evidence package is similar to 510(k) but more extensive, because you're defining the performance standards for a new device type rather than comparing to an existing one. Clinical data requirements are typically higher. The special controls you propose — performance testing requirements, labelling requirements, post-market surveillance requirements — become the standard for the category.
Timeline and cost
FDA's review target for De Novo is 150 days. Total program time including AI cycles typically runs 12–18 months from submission. FY2026 user fees are approximately $170,000 standard or $42,000 small business. Total program cost, including consulting and clinical evidence work, typically runs €1M–€3M+.
The strategic value of De Novo
If your De Novo request is granted, you become the predicate for your device category. Competitors entering the same category can reference your cleared device in their 510(k)s — which is a market signal, not a disadvantage, because it confirms you've defined the category.
Being first to De Novo in a clinical AI category is a regulatory moat. The 12–18 months it takes to authorise the category is the same 12–18 months your competitors lose if they have to take their own De Novo path behind you.
Pathway 3: PMA — Premarket Approval
PMA is required for Class III devices — those that support or sustain life, or present a potentially unreasonable risk of illness or injury. It's the most rigorous pathway and requires valid scientific evidence — typically including randomised controlled trial data — to demonstrate reasonable assurance of safety and effectiveness.
When AI medical devices require PMA: automated diagnosis systems that replace, rather than inform, clinical judgement; AI systems that directly control or recommend treatment in high-risk clinical scenarios; novel device types where FDA determines the risk level warrants Class III classification.
Most AI diagnostic decision-support tools — which flag findings for clinical review rather than replacing clinical review — do not require PMA. The key determinant is whether the device's output directly drives a clinical action without clinician interpretation.
Timeline and cost
PMA is a multi-year process. Plan 3–5 years from study design to approval, depending on the clinical category and the pace of data collection. FY2026 user fees are approximately $540,000 standard (with additional fees for amendments and supplements), or roughly $135,000 for small business — a 75% reduction.
Historically, PMA programs for novel AI devices have been preceded by 5–7 year programs at established device manufacturers, not led by startups. PMA is not typically the right pathway for a Series A or B AI company without late-stage institutional partner backing.
The Predetermined Change Control Plan (PCCP): the AI overlay
The standard FDA regulatory framework assumes that a cleared device remains substantially the same as what was cleared. For AI/ML devices, this creates a structural problem: the value of AI comes partly from its ability to improve over time through retraining. But every significant change to an AI/ML device may otherwise require a new premarket submission.
FDA's response is the Predetermined Change Control Plan (PCCP), introduced in the January 2021 AI/ML Action Plan, formalised through draft guidance in April 2023, and finalised in December 2024 ("Predetermined Change Control Plans for Machine Learning-Enabled Device Software Functions"). PCCPs are explicitly applicable across all three pathways — 510(k), De Novo, and PMA.
A PCCP is a document submitted as part of your premarket submission that pre-specifies the types of modifications you anticipate making to your AI/ML device; the methodology for implementing those modifications; the performance testing and validation you will conduct for each modification type; and the criteria that would trigger a new premarket submission rather than deployment under the PCCP.
If FDA approves your PCCP as part of your clearance or authorisation, you can implement the pre-specified modifications without submitting a new 510(k) or De Novo — as long as the modifications stay within the approved PCCP scope and you conduct the pre-specified validation.
For AI device manufacturers planning ongoing model improvement, a PCCP is not optional — it's the framework that allows you to operate compliantly after clearance. If you're operating a locked model that you don't plan to retrain, a PCCP isn't required. Most serious AI programs aren't in that camp, and drafting the PCCP as an afterthought generates either a weak PCCP that doesn't cover the modifications you actually want to make, or a PCCP that FDA won't approve because it's too broad.
The Pre-Submission programme (Q-Subs)
Before formal submission, FDA's Pre-Submission (Q-Submission) programme lets you request structured feedback on your regulatory strategy. Several Q-Sub types exist: Pre-Submission for feedback on the planned approach, Submission Issue Request for issues on an in-review submission, Study Risk Determination for IDE clarity, and Informational Meeting for higher-level discussions.
Typical turnaround is 70–90 days from request to written response. Meetings, where offered, run 60–90 minutes. The Q-Sub minutes become part of the regulatory record — what FDA tells you in a Pre-Submission isn't binding, but it documents the agency's contemporaneous view and is materially valuable later if disagreements arise.
For founders pursuing a US strategy for the first time, a Pre-Sub before finalising your pathway is one of the highest-ROI 90-day waits available. FDA will give you informal feedback on intended classification and pathway selection that significantly de-risks the submission decision.
The Breakthrough Device Program
FDA's Breakthrough Device Program provides more interactive FDA engagement and prioritised review for devices that provide more effective treatment or diagnosis of serious conditions. AI diagnostics frequently qualify — over 80 AI/ML-enabled devices have received Breakthrough designation as of 2024. Designation provides "Sprint" discussions for time-bounded resolution of specific regulatory questions and earlier FDA input on study design.
Breakthrough designation doesn't change the pathway — you still need 510(k), De Novo, or PMA — but it materially accelerates the review and shapes the evidence requirements toward what FDA actually wants to see. Worth applying for if your device addresses a serious or life-threatening condition with unmet clinical need.
EU MDR vs FDA: what transfers and what doesn't
For companies pursuing both EU and US clearance — which is most European AI health startups at Series B — the temptation is to assume that EU MDR experience translates directly to FDA strategy. It does, partially.
What transfers
Clinical validation methodology — retrospective dataset analysis, prospective study design, performance metric selection — is largely aligned between EU MDR and FDA. IEC 62304 software lifecycle documentation is required in both jurisdictions. ISO 14971 risk management is referenced by both frameworks. IEC 62366-1 usability evaluation is applicable to both. The principle of intended use specificity applies in both. Cybersecurity documentation aligns substantially between FDA's September 2023 guidance and IEC 81001-5-1 (used under EU MDR).
What doesn't transfer
The EU notified body review process has no FDA equivalent; FDA conducts its own submission review directly, with Advisory Committee input for higher-risk devices. EU MDR's classification Rule 11 doesn't map cleanly to FDA Class I/II/III — the same device may land in different risk classes under each system. The clinical evidence standard differs: FDA is generally more prescriptive about study design requirements, particularly for novel device types where De Novo special controls effectively define the evidence package. Post-market surveillance requirements differ significantly: EU MDR's PMCF obligations and FDA's post-market surveillance requirements have different triggers and different scope. The PCCP has no direct EU MDR equivalent, though the AI Act's change-management provisions are evolving toward similar principles.
The EU AI Act layer (EU-bound programs only)
For programs targeting EU launch in 2026 or later, the EU AI Act sits on top of MDR/IVDR. AI-enabled medical device software regulated as MDR Class IIa or above is automatically classified as high-risk under the AI Act, with conformity assessment integrated into the MDR route but adding documentation on training data governance, transparency to deployers, human oversight, and AI-specific post-market monitoring. High-risk provisions phase in from August 2026. US-only programs don't face this. Programs pursuing both jurisdictions need to plan the AI Act layer as a parallel workstream from week one. (Our six-month roadmap piece covers AI Act integration alongside MDR in detail.)
The practical implication: EU MDR and FDA programmes can share clinical data, software documentation, cybersecurity work, and usability evaluation. The regulatory strategy, submission format, and evidence framing need to be built independently for each jurisdiction.
Post-market reporting and surveillance
Clearance is the start of a regulatory relationship, not the end. FDA-cleared AI medical devices face ongoing post-market obligations:
Medical Device Reporting (MDR) — mandatory reporting of adverse events and certain product malfunctions to FDA, with specific timelines (5-day, 30-day depending on severity).
Post-approval studies — typically required for PMA-approved devices and sometimes for De Novo-authorised devices, designed to capture real-world performance over time.
PCCP-related performance monitoring — if your clearance included a PCCP, monitoring against the pre-specified validation criteria is required for changes deployed under the PCCP.
Cybersecurity vigilance — ongoing vulnerability monitoring, patching, and disclosure under FDA's September 2023 cybersecurity guidance.
How to decide: a practical framework
Three questions determine your pathway, applied in order.
1. Does a substantially equivalent predicate exist? Search FDA's 510(k) database (accessible at fda.gov/medical-devices) for cleared devices in your clinical category with a comparable intended use. If a clear predicate exists with similar technology and indications, 510(k) is your pathway. If the closest predicate has a meaningfully different intended use or technology, assess whether the substantial equivalence argument is defensible before committing to 510(k). FDA's AI/ML-enabled medical device list is the right starting point for AI-specific predicates.
2. What is the risk level of your device's intended use? If your device directly controls a clinical intervention or makes autonomous treatment decisions, Class III and PMA may be unavoidable. If your device informs clinical decision-making (a clinician reviews and acts on the output), Class II and 510(k)/De Novo is almost certainly the right target.
3. Do you need to establish the category? If no AI device has been cleared in your clinical area and you have the resources for an 18-month De Novo program, the regulatory moat from being the category-defining predicate may be worth the additional timeline. If speed to market is the priority and a reasonable predicate exists, 510(k) with a strong predicate argument is faster.
For founders, the practical implication is that post-market infrastructure — adverse event tracking, performance monitoring, customer support and complaint handling — needs to be designed before clearance, not bolted on after.
Where Vector Labs fits
We work with AI medical device founders on US regulatory strategy at three points: pathway selection (classification, predicate analysis, build/buy/partner decisions for US market entry), Pre-Submission strategy and execution (Q-Sub preparation, FDA engagement, response to feedback), and submission preparation (510(k) or De Novo file construction, PCCP design, post-market infrastructure).
If you're scoping a US regulatory strategy — particularly alongside an EU MDR programme — and want to pressure-test the plan, get in touch at vector-labs.ai.
For the broader series: SaMD classification covers the threshold question of whether your software is regulated at all; the six-month roadmap covers EU regulatory program execution with direct parallels to FDA program planning; the engineering reality of clinical-grade AI covers the technical evidence package; the business case for clinical AI covers the strategic frame around the regulatory work.