



Класификация 101 по SaMD: Как да определите дали вашият алгоритъм е медицинско изделие, преди регулаторът да го направи


Най-скъпата регулаторна грешка, която един стартъп в областта на здравния изкуствен интелект може да направи, е да открие късно, че алгоритъмът му е медицинско устройство.
Не защото е катастрофално да си медицинско изделие. Не е. Много печеливши, добре финансирани компании за здравен изкуствен интелект работят като регулирани производители на медицински изделия. Проблемът е, че се открива късно: след като сте изградили архитектура, събрали данни, подписали клинични партньорства и понякога сте доставили на потребителите без контрола върху дизайна, документацията и инфраструктурата за валидиране, които софтуерът на медицинското изделие изисква. Модернизирането им върху съществуващ продукт е значително по-трудно и по-скъпо от вграждането им от самото начало. Виждали сме, че модернизациите поглъщат 12+ месеца и няколко милиона евро допълнителни разходи за програми, които щяха да бъдат рутинни, ако класификацията беше уредена още през първия месец.
Тази статия е за това как да вземете това решение рано, преди първото си клинично партньорство, преди вашата Серия А и в идеалния случай преди да напишете много код. Рамките съществуват. Анализът е изпълним. Това, което липсва на повечето основатели, е ясно обяснение как да ги прилагат.
Това е допълнение към нашите статии за изграждане на сърдечен изкуствен интелект с клинично ниво — от инженерна и бизнес гледна точка . След като определите, че сте медицинско устройство, тези статии описват какво следва.
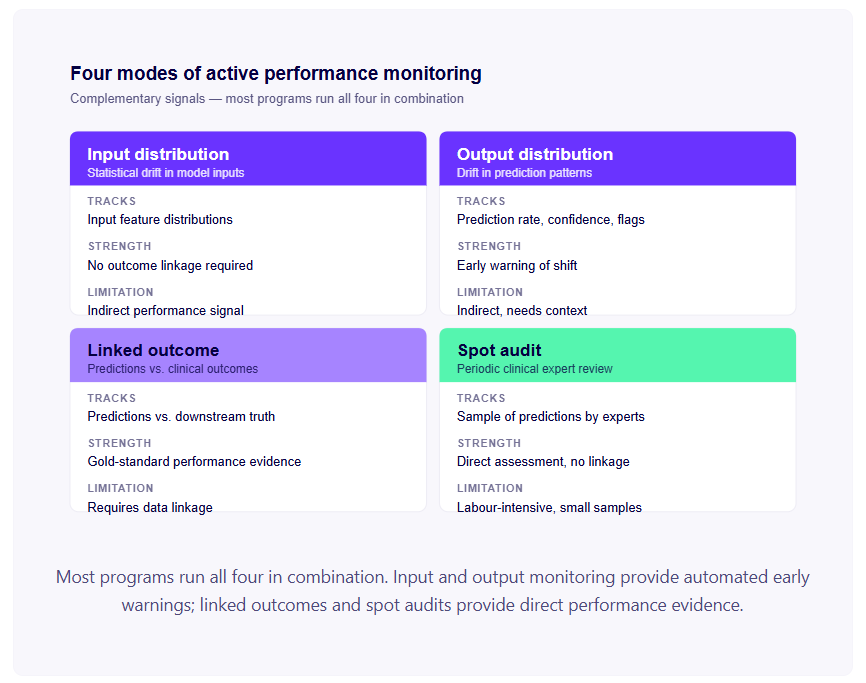
Защо „медицинско устройство“ се отнася за повече софтуер, отколкото си мислите
Основателите често приемат, че „медицинско устройство“ означава физически хардуер, като например ЕКГ монитор, инфузионна помпа, хирургически робот. Софтуерът, който просто обработва здравни данни, показва информация или поддържа клинични работни процеси, се намира в сива зона, предполагат те.
Това предположение е погрешно в ЕС, САЩ, Обединеното кралство и повечето други големи регулаторни юрисдикции.
Всички основни западни регулатори имат изрични рамки за Софтуер като медицинско устройство (SaMD) - софтуер, който изпълнява една или повече медицински цели независимо от хардуерно медицинско устройство. Международният форум на регулаторите на медицинските изделия (IMDRF), чието определение за SaMD е в основата на регулаторите в САЩ, ЕС, Великобритания, Канада, Австралия, Сингапур и Япония, определя SaMD като „софтуер, предназначен да се използва за една или повече медицински цели, който изпълнява тези цели, без да е част от хардуерно медицинско устройство“.
Ако вашият алгоритъм е предназначен да диагностицира състояние, да прогнозира резултата за пациента, да препоръча лечение, да наблюдава състоянието на пациента или да открие заболяване, той почти сигурно е медицинско устройство, независимо дали работи на телефон, уеб сървър или болнична работна станция.
Архитектурните решения не променят това. Отказите от отговорност не променят това. Фактът, че „клиницистът взема окончателното решение“, не променя това. Класификацията се определя от предназначението на вашия софтуер, а не от начина, по който го структурирате.
Рамката IMDRF SaMD: първият филтър
Класификационната рамка на IMDRF SaMD (IMDRF/SaMD WG/N12FINAL) класифицира софтуера по две измерения:
Значение на предоставената информация за решението за здравеопазване: лечение или диагностика, насочване на клиничното управление или информиране на клиничното управление.
Състояние на здравната ситуация: критично (животозастрашаващо или критично във времето), сериозно или несериозно.
Матрицата произвежда четири категории SaMD:
Категория IV е с най-висок риск; Категория I е с най-нисък. Рамката е важна, защото е отправната точка, която FDA и регулаторните органи на ЕС използват, когато оценяват кои контролни механизми са подходящи, и защото тя предвижда къде ще се озове устройството ви, преди да подадете каквото и да било.
Няколко примера, описани в регулаторните режими, следват:
Алгоритъм, който интерпретира 12-канална ЕКГ в спешно отделение, сигнализирайки за съмнение за STEMI за незабавно активиране на катетеризационна лаборатория: Лечение/Диагноза × Критично = Категория IV . Вероятен EU MDR Клас IIb (решенията могат да доведат до сериозно влошаване, ако бъдат пропуснати); FDA 510(k) или De Novo в зависимост от наличието на предикат.
Алгоритъм, който анализира изображения на ретината, за да открие диабетна ретинопатия и ги маркира за офталмологичен преглед: Лечение/Диагностика × Сериозно = Категория III . Обикновено клас IIa по MDR на ЕС; път De Novo на FDA (пътят, поет от IDx-DR).
Функция за откриване на предсърдно мъждене (AFib) на носимо устройство, която предупреждава потребителя да се консултира със здравен специалист, ако се открие нередовен ритъм: Inform × Serious = Категория I по IMDRF, въпреки че обикновено все още е клас IIa по EU MDR и клас II по FDA по De Novo (пътят, по който е поела функцията за предсърдно мъждене на Apple Watch). Рамката на IMDRF е таксономия на риска; класовете MDR и FDA са регулаторни пътища. Те се припокриват, но не се равняват.
30-дневна оценка на риска от повторно приемане в болница се появи на клинично табло: Информация × Сериозно = Категория I съгласно IMDRF, но вероятно EU MDR Клас IIa съгласно Правило 11.
Алгоритъм, който предлага хранителни препоръки в неклинично приложение за уелнес: Информация × Несериозно = Категория I съгласно IMDRF и извън регулацията на медицинските изделия изцяло съгласно общата политика за уелнес на FDA, ако не са направени конкретни твърдения за заболяване.
Изводът: IMDRF е първият ви филтър за обмисляне на риска, но не определя вашия регулаторен път. Правилата, специфични за юрисдикцията по-долу, го правят.
ЕС MDR: анализ на Правило 11
Съгласно EU MDR, класификацията на софтуера се регулира предимно от Правило 11 в Приложение VIII. Действителната структура на правилата за софтуер, предоставящ информация, използвана при клинични решения:
Клас IIa (по подразбиране): Софтуер, предоставящ информация, използвана за вземане на решения с диагностична или терапевтична цел. Повечето софтуер за подпомагане на вземането на решения с изкуствен интелект попада тук.
Клас IIb: Когато решенията биха могли да причинят сериозно влошаване на здравословното състояние на дадено лице или хирургическа интервенция .
Клас III: Когато решенията биха могли да причинят смърт или необратимо влошаване на здравословното състояние на дадено лице .
Отделно подправило обхваща софтуера за мониторинг:
Клас IIa (по подразбиране за мониторинг): Софтуер, предназначен за наблюдение на физиологични процеси.
Клас IIb (мониторинг): Софтуер за наблюдение на жизненоважни физиологични параметри, чието отклонение може да доведе до непосредствена опасност.
Софтуерът, който не попада в нито една от тези категории, е клас I. Съгласно Правило 11, клас I рядко се среща на практика за здравен ИИ; повечето истински диагностичен или терапевтичен софтуер, подпомагащ вземането на решения, попада в IIa или IIb.
Работени приложения:
Система с изкуствен интелект, която сигнализира за потенциални сърдечни аномалии за преглед от кардиолог: Клас IIa . Решенията се преглеждат от клиницист; последицата от грешка е сериозна, но не причинява пряко смърт или необратимо влошаване на клиничния цикъл.
Система с изкуствен интелект, която прогнозира риска от сепсис и го показва на клинично табло: обикновено клас IIb . Информацията е в основата на решенията, чието последствие (пропуснат сепсис) може да бъде сериозно влошаване или смърт.
Система с изкуствен интелект, която автоматично регулира доставянето на инсулин въз основа на мониторинг на глюкозата: Клас III съгласно по-широките правила на MDR, тъй като директно задвижва лечение с непосредствени опасни последици.
Анализът на „потенциалните последици“ е мястото, където възникват повечето спорове относно класификацията. Последиците зависят от клиничния контекст, спешността и дали клиницистът преглежда резултата преди действие. Прецизното документиране на предвидената употреба – уточняване, че резултатите се преглеждат от квалифицирани клиницисти, както и уточняване на клиничния контекст – може да промени анализа на последствията и по този начин класификацията.
За наистина гранични продукти съществуват три формални механизма: граничните ръководства на MDCG (Координационна група за медицински изделия), писмено становище за класификация от нотифициран орган или взаимодействие с компетентен орган преди подаване на заявление. Това са формални регулаторни канали, а не неформални консултации, и са значително по-евтини от откриването по средата на проекта, че класификацията ви е била грешна.
Закон на ЕС за изкуствения интелект: наслагването, което добавя втори регулаторен режим
За продуктите с изкуствен интелект в здравеопазването в ЕС, Законът за изкуствен интелект (ИИ) вече е над Регламента за медицинските продукти (MDR). Към май 2026 г., с постепенното въвеждане на разпоредбите на Закона за ИИ с висок риск от август 2026 г., това е най-големият активен източник на объркване сред основателите в регулирането на ИИ в здравеопазването.
Структурата: софтуерът за медицински изделия, регулиран като клас IIa, IIb или III съгласно MDR , автоматично се класифицира като високорисков съгласно Закона за изкуствения интелект (член 6, във връзка с приложение I, обхващащо устройства, които изискват оценка на съответствието от трета страна). Няма избор на дизайн, който да избягва класификацията като високорисков за регулиран медицински изделия с изкуствен интелект.
Какво добавя класификацията за висок риск, в допълнение към MDR:
Управление на документираните данни за обучение — качество на данните, представителност, тестване за пристрастност, демонстриране, че данните са подходящи за предвидената употреба.
Разпоредби за прозрачност за внедрителите (болници, клиники) относно възможностите, ограниченията, очакваната точност и предназначението, достатъчни за безопасна експлоатация.
Механизми за човешки надзор, документирани в дизайна — за SaMD, работещи с клиницисти, това е лесно; за автономни системи за вземане на решения това е смислено ограничение на дизайна.
Следпазарно наблюдение на системата с изкуствен интелект, което се припокрива, но е различно от следпазарното наблюдение на MDR.
Оценка на съответствието на системата с изкуствен интелект. За SaMD, която вече изисква участието на нотифициран орган съгласно MDR, оценката на съответствието по Закона за изкуствения интелект е до голяма степен интегрирана в съществуващия процес; документалната тежест е реална, но процедурният път съществува.
Стратегическото значение: всяка програма за здравен изкуствен интелект, насочена към стартиране в ЕС през 2026 г. или по-късно, трябва да планира спазването на Закона за изкуствения интелект като паралелен работен поток от първия ден, а не да го преосмисля след MDR. Програмите, които вече са преминали през оценка на съответствието с MDR, имат значително предимство пред новите участници, които трябва да преминат и двата режима едновременно.
FDA: регулиране на софтуерните функции и изключение от CDS
Подходът на FDA към класификацията на SaMD приблизително съответства на IMDRF, но е оперативно различен. FDA използва насоки за функциите на софтуера на устройството, за да определи кои функции са регулирани като устройства и кои не подлежат на надзор.
Софтуер FDA не регулира като медицинско изделие: административен софтуер (планиране, фактуриране, комуникация); софтуер за общо здраве, който не дава специфични здравни препоръки; електронни досиета на пациенти, непредназначени за подпомагане на клиничните решения; софтуер, който прехвърля, съхранява или показва клинични лабораторни резултати, без да ги интерпретира.
Софтуер FDA регулира: софтуер, който анализира специфични за пациента данни, за да направи или подпомогне диагностична или лечебна препоръка; софтуер, предоставящ специфични за пациента предупреждения въз основа на клинични данни; софтуер, извършващ анализ на изображения за диагностични цели; софтуер, анализиращ физиологични сигнали за клинични цели.
Изключението за клинична подкрепа при вземането на решения (CDS) — кодифицирано в Закона за лечението през 21-ви век, въведено чрез окончателното ръководство на FDA за CDS (септември 2022 г.) — е частта от рамката, която основателите на FDA най-често неправилно тълкуват. Четирите критерия, и четирите от които трябва да бъдат изпълнени , са:
- Софтуерът не придобива, обработва или анализира медицинско изображение, сигнал от ин витро диагностично устройство или модел/сигнал от система за събиране на сигнали.
- Софтуерът показва, анализира или отпечатва медицинска информация за пациент или група пациенти.
- Софтуерът предоставя препоръки на медицински специалист, а не директно на пациент.
- Софтуерът предоставя основата за препоръката по начин, който позволява на медицинския специалист да я прегледа самостоятелно, а не да разчита предимно на нея.
Критерий (1) е мястото, където повечето медицински софтуер с изкуствен интелект не отговарят на изискванията за изключение, независимо от това колко прозрачен е моделът. Ако анализирате ЕКГ сигнали, изображения на ретината, дерматологични снимки, патологични слайдове, ултразвук, ЯМР или друг сигнал от система за медицинско събиране на сигнали, нямате право на изключение за CDS, които не са за устройства. Точка. Прозрачността, обяснимостта и клиничната ангажираност нямат значение за критерий (1); самият начин на въвеждане ви дисквалифицира.
Критерият за прозрачност (критерий 4) е този, върху който се фокусират основателите, и той има значение - но само за CDS, които не са свързани с изображения и сигнали. Обработка на клинични бележки на естествен език, оценяване на риска върху структурирани данни от електронните здравни заведения, системи за предупреждение, изградени върху лабораторни стойности, които не сте придобили сами: за тях обясним модел, който разкрива своите разсъждения, е значително по-лесен за поддържане извън регулирането на FDA, отколкото модел, представляващ черна кутия. За ИИ на изображения и сигнали, фокусирайте енергията си другаде.
За основателите, които изграждат изкуствен интелект, базиран на изображения или сигнали, реалистичният път от FDA е одобрение 510(k), ако съществува съществено еквивалентно предикатно устройство, или класификация De Novo, ако такова не съществува. И двата са пътища за устройства, а не изключения.
IVDR: другият път, по който може би сте поели, без да го знаете
Някои софтуерни продукти са регулирани от Регламента на ЕС за инвитро диагностика (IVDR, 2017/746) , а не от MDR. Това изненадва основателите.
IVDR се отнася за софтуер, който интерпретира резултатите от in vitro диагностичен тест - лабораторни резултати, данни за геномно секвениране, измервания на биомаркери, анализ на микробиом, резултати от имуноанализ. Ако основната функция на вашия софтуер е да интерпретира данни от проба, взета от човешкото тяло и обработена in vitro, вероятно сте IVDR продукт.
IVDR има собствена система за класификация (класове A, B, C, D) съгласно различни правила от MDR Правило 11. Устройствата от клас C и D подлежат на участието на нотифициран орган. Устройствата от клас B подлежат на оценка на съответствието от нотифициран орган за някои аспекти. Клас A е с най-нисък риск. Много продукти с изкуствен интелект, интерпретиращи геномни или биомаркерни данни, попадат в клас C, което носи значителна тежест за оценка на съответствието.
Основателите, които разработват геномен ИИ, ИИ за биомаркери, ИИ за микробиоми или ИИ за интерпретация на лабораторни резултати, трябва да ги класифицират като IVDR, а не като MDR. Изискванията за техническа и клинична оценка се припокриват съществено, но правилата и пътищата за съответствие са различни.
Американските основатели, които разработват софтуер за IVD, са изправени пред регулации на FDA съгласно 21 CFR 809, както и съответната класификация на устройствата (клинична химия, имунология, хематология), с припокриващи се принципи на SaMD. IVD продуктите, базирани на изкуствен интелект, все по-често преминават през De Novo или 510(k).
UKCA, MHRA и продължаващият преход в Обединеното кралство
За стартиращи компании, базирани в Обединеното кралство, Агенцията за регулиране на лекарствата и здравните продукти (MHRA) прилага поетапен преход от запазените правила на ЕС MDR/IVDR. Позицията се е променяла многократно след Brexit и всяка конкретна дата във всяко обяснение (включително това) трябва да бъде проверена спрямо публикуваната в момента пътна карта на MHRA, преди да се разчита на нея.
Настоящо състояние: Устройствата с маркировка „CE“ продължават да се приемат във Великобритания за определени периоди, като сроковете за постепенно премахване варират в зависимост от класа на устройството. Новите разпоредби в Обединеното кралство се въвеждат на етапи през 2025–2027 г., като разпоредбите за следпускане на пазара и основните разпоредби влизат в сила на различни етапи.
Практическото значение за основателите: изграждане на основната техническа документация — клинична оценка, система за управление на качеството по ISO 13485, софтуерна документация по IEC 62304, управление на риска по ISO 14971, използваемост по IEC 62366-1 — по стандарт, който удовлетворява както изискванията на ЕС за медицински продукти (MDR), така и изискванията на Обединеното кралство за медицински продукти (MDR). Същността е до голяма степен съгласувана. Пътищата за оценка на съответствието (нотифицирани органи в ЕС срещу одобрени органи в Обединеното кралство) са отделни, но основният файл е един и същ. За стартиращи компании с търговски амбиции в NHS съответствието в Обединеното кралство е предпоставка; за търговски амбиции в ЕС, маркировката CE е пътят; и за двете работата по документацията е паралелна, а не дублираща се.
MHRA също така управлява програмата AI Airlock — регулаторна пясъчна кутия, специално предназначена за медицински устройства с изкуствен интелект, полезна за продукти в ранен етап на разработка, при които има неясна класификация или нови предназначения. Законът на ЕС за изкуствен интелект (ИИ) налага регулаторни пясъчници за ИИ на ниво държави членки. За истински здравен ИИ в ранен етап в сиви зони, пясъчните кутии са недостатъчно използван вариант.
Сивата зона: къде основателите грешат
Три категории софтуер постоянно се класифицират погрешно от основателите.
„Това просто показва информация на лекаря, а не вземане на решение.“ Както рамките на EU MDR, така и на FDA регулират софтуера, който предоставя информация, използвана при вземането на клинични решения – не само софтуер, който взема автоматизирани решения. Ако вашият алгоритъм анализира данните за пациента и извежда оценка на риска, вероятност, флаг или класиран списък, който клиницистът след това използва, за да вземе клинично решение, вие предоставяте информация, използвана при вземането на решения. Вероятно сте медицинско изделие.
„Това е приложение за уелнес, а не клиничен инструмент.“ Регулаторната граница между уелнес и клинично се определя от заявената и разумно предвидима употреба на софтуера, а не от това, как го нарича разработчикът. Ако вашият алгоритъм за „уелнес“ анализира физиологични данни, за да сигнализира за потенциално сериозно състояние – дори неформално, дори с отказ от отговорност – регулаторните органи разглеждат действителната функция, а не етикета.
Отказите от отговорност, които гласят „това не е медицински съвет“, не променят регулаторния статус на софтуера, който изпълнява функции на медицинско устройство. Това е най-скъпоструващото погрешно схващане в ранния етап на здравен изкуствен интелект.
Политиката на FDA за общо здраве предоставя тясна безопасна зона за софтуер, който поддържа или насърчава общото здраве (проследяване на съня, насърчаване на фитнеса, управление на стреса), без да прави конкретни твърдения за заболяване. В момента, в който вашият „уелнес“ продукт направи някакво конкретно твърдение за откриване на заболяване или риск, безопасната зона се изпарява.
„Ние сме само за изследователска употреба (RUO) или експериментална употреба.“ RUO е специфично изключение за етикетиране от FDA за in vitro диагностични продукти в предварителните пазарни проучвания, а не обща защита за SaMD. За софтуера в по-широк смисъл, съответните предварителни пазарни пътища са Изключението за изследователски изделия (IDE) на FDA за клинични изпитвания или разпоредбите на ЕС за клинични изпитвания съгласно MDR.
Във всички случаи защитата е обвързана с действителна употреба за научни изследвания. Ако инструмент с етикет RUO или етикет за изследователски цели се използва в клиничната практика – дори неофициално, дори без изрично рекламиране – защитата се изпарява. FDA е предприела действия срещу компании, чийто софтуер с етикет RUO е бил използван клинично. Партньорствата с NHS и пилотните болнични проекти, които се насочват към рутинна клинична употреба, са особено често срещан начин за загуба на защита за изследователски цели, без да се осъзнава това.
Последици от разходите и времевата рамка по клас
Приблизителна представа за това колко струва всяка класификация във време и пари за типичен стартъп в областта на здравния изкуствен интелект, извлечена от програми, с които сме работили:
FDA CDS (системни системи за контрол на качеството на живот), които не са свързани с устройства, или общо здраве: Не се изисква подаване на заявление до FDA. Време за пускане на пазара: седмици до месеци. Регулаторни разходи: ниски — правно становище, документиран анализ на предназначението. Уловката: критериите са тесни и изкуственият интелект с изображения/сигнали не отговаря на условията.
EU MDR Клас I: Самостоятелна декларация за съответствие, без участието на нотифициран орган за повечето продукти от клас I. Общо регулаторни усилия: 6–12 месеца и 50 000–200 000 евро. Все по-рядко срещано за здравния ИИ съгласно Правило 11.
EU MDR Клас IIa (повечето диагностични решения, подпомагащи ИИ): Участие на нотифициран орган. Общо регулаторни усилия от сериозно начало: 12–24 месеца и 300 000–800 000 евро, плюс 5–10 човеко-години писане на регулаторни документи и клинична оценка. Само таксите на нотифицирания орган често са 100 000–300 000 евро.
EU MDR Class IIb / III (подкрепа за вземане на решения при висок риск, автономно лечение): 24–48 месеца и 1–5 милиона евро+ в зависимост от обхвата на клиничната оценка и дали са необходими нови клинични изследвания.
FDA 510(k) (одобрение въз основа на предикати): 9–18 месеца от подаване, плюс 6–18 месеца подготовка. 300 000–1 милион+ евро в зависимост от нуждите от клинични данни. Достъпно само ако съществува съществено еквивалентно предикатно устройство.
FDA De Novo (без предикат): 12–24 месеца от подаването, плюс обширна подготовка. 1–3 милиона евро+. Първите по рода си продукти за сърдечен изкуствен интелект обикновено преминават през De Novo.
Обвързващо ограничение, което много основатели пропускат: капацитетът на нотифицираните органи в Европа в момента е истинско пречка. Ангажирането на нотифициран орган и спазването на графика му трябва да се извършва преди да ви е необходим, а не след това. Някои категории устройства в момента имат чакане от 12+ месеца само за да започне процесът на оценяване на съответствието.
Тези числа са причината класификацията да е важна. Разликата между Клас I и Клас IIa е приблизително една година и няколкостотин хиляди евро. Разликата между Клас IIa и Клас IIb е приблизително още една година и още няколкостотин хиляди. Грешното класифициране в бизнес плана ви е сериозна грешка – и „ще го разберем в Серия А“ не е план.
Защита на данните: GDPR и HIPAA са начело на всичко
Класификацията на медицинските изделия и съответствието със защитата на данните са отделни, паралелни регулаторни режими. Да бъдеш медицинско изделие от клас IIa в ЕС не те освобождава от GDPR. Да бъдеш изделие, одобрено от FDA в САЩ, не те освобождава от HIPAA. Двата режима имат припокриващи се, но различни изисквания – и тежестта на защитата на данните често надвишава тежестта на документирането на медицинското изделие в суров вид.
Здравните данни имат строги разпоредби и в двете юрисдикции (Член 9 от GDPR; Правилата за поверителност и сигурност на HIPAA), а изискванията за управление на данните за обучение по Закона за изкуствения интелект взаимодействат с GDPR по начини, които все още се изясняват. Законодателството на САЩ на щатско ниво (CMIA на Калифорния, Законът „Моето здраве, моите данни“ на Вашингтон и други) добавя допълнителна сложност. Основател, който изгражда здравен изкуствен интелект, работи при поне три регулаторни режима – медицински изделия, ИИ/защита на данните и специфично за юрисдикцията законодателство за здравните данни – които се нуждаят от интегриран дизайн за съответствие.
Как да се вземе решението: практичен процес
Стъпка 1: Документирайте предназначението си. Опишете точно какво е предназначен вашият софтуер, за кого, в какъв клиничен контекст и с какви очаквани последици от клиничното решение. Бъдете конкретни. „Изкуствен интелект, който помага на клиницистите“ не е предназначение. „Софтуер, който анализира 12-канални ЕКГ записи, за да идентифицира модели, съответстващи на предсърдно мъждене, и показва маркирани записи за преглед от кардиолог в кардиологична амбулаторна обстановка“ е предназначение. Този документ е отправната точка на всеки регулаторен анализ, който някога ще правите, а прецизното му ранно извършване спестява огромна работа впоследствие.
Стъпка 2: Приложете рамката IMDRF. Въз основа на предназначението си определете вашата категория SaMD (I–IV). Това ви дава предварително ниво на риск.
Стъпка 3: Приложете правилата за класификация на съответната юрисдикция. За ЕС MDR: приложете Правило 11 (и проверете дали се прилага IVDR, ако обработвате изходни данни от IVD). За САЩ: приложете ръководството на FDA за софтуерните функции и четирикритериалния тест за CDS, които не са за устройства. За Обединеното кралство: приложете текущата позиция на Обединеното кралство по MDR (в значителна степен съгласувана с ЕС MDR, но проверете пътната карта на MHRA). Приложете наслагването на Закона за изкуствения интелект за всеки продукт, обвързан с ЕС. Документирайте анализа писмено.
Стъпка 4: Ако сте в сива зона, получете официално становище. Съществуват три формални механизма: среща преди подаване с FDA (програма Q-Submission) , искане за научна консултация от MHRA или становище за класификация от нотифициран орган на ЕС (с позоваване на граничните насоки на MDCG). Това не са неформални консултации; те са формални регулаторни канали с документирани резултати. За истински сиви зони официалното становище е значително по-евтино от откриването по средата на проекта, че вашият анализ на класификацията е бил грешен.
Стъпка 5: Обмислете програми с пясъчници за ранна несигурност. Регулаторните пясъчници на MHRA AI Airlock и Закона за изкуствения интелект на ЕС предоставят структурирана среда за развитие на здравен изкуствен интелект в ранен етап, заедно с регулаторните органи. Те се използват недостатъчно, особено от основатели със седалище в САЩ, които не осъзнават, че са опция.
Стъпка 6: Проектирайте системата си за определената от вас класификация. Ако сте производител на медицинско изделие, започнете да изграждате СУК, да документирате входните и изходните данни от проекта и да внедрявате управление на риска сега, а не когато сте готови да го представите. Цената за преоборудване на контролни механизми за проектиране върху съществуващ продукт, според нашия опит, е приблизително 3–5 пъти по-висока от цената за изграждането им от самото начало, а сроковете обикновено са 6–18 месеца. Програмите, които третират регулаторната инфраструктура като проблем от Серия Б, са програми, които не достигат Серия Б.
Къде е мястото на Vector Labs
Работим с основатели на здравни компании в областта на изкуствения интелект и медицински технологични компании по отношение на регулаторната стратегия в три аспекта: определяне на обхвата на класификацията (определяне къде ще се наложи вашият продукт и какви са регулаторните последици), регулаторна проверка (оценка на програми в процес на разработка или цели за придобиване за регулаторно излагане) и инженерно изпълнение (изграждане на система за управление на качеството (QMS), контрол на дизайна, техническо досие и инфраструктура за клинична оценка, които се изискват за маркировка CE или подаване на документи до FDA).
Ако сте в ранен етап и не сте сигурни дали вашият алгоритъм е медицинско изделие, това е най-евтиният момент да получите правилния отговор. Свържете се с нас на vector-labs.ai .
За минали класификации на основателите — всъщност изграждащи сърдечен ИИ до клинично ниво — вижте нашите съпътстващи статии за инженерната реалност и бизнес казуса . Заедно трите статии обхващат пътя от „медицинско изделие ли е моят алгоритъм?“ до „как да доставя продукт с маркировка CE и да изградя защитим бизнес с сърдечен ИИ?“.