



Следпазарно наблюдение за медицински изделия с изкуствен интелект: Защо маркировката CE е началото, а не краят


Маркировката CE е важен етап. Тя не е крайна точка.
За повечето традиционни медицински изделия, следпродажбеното наблюдение е систематична, но сравнително пасивна дейност — събиране на данни за оплаквания, наблюдение на нежелани събития, преглед на литературата и периодично актуализиране на клиничната оценка. За медицинските изделия с изкуствен интелект, следпродажбеното наблюдение е фундаментално активна и непрекъсната програма — защото връзката между вашето устройство и реалния свят се променя с времето по начини, които традиционните устройства не изпитват.
Разпределението на данните, генерирани от клиничната практика, се променя. Популациите пациенти се променят. Оборудването за образна диагностика се обновява. Клиничните работни процеси се развиват. Моделът, валидиран през 2024 г. върху данни от три клинични центъра, може да се представи съществено различно през 2026 г. върху данни от двадесет клинични центъра, обслужващи различни групи пациенти с различно оборудване.
Това не е хипотетичен риск. Публикуваните изследвания документират спад в производителността от 10–20 процентни пункта, когато диагностичните системи с изкуствен интелект се оценяват върху външни набори от данни, които не са представени в обучението. Екстраполирайки към разнообразието в реалното внедряване и многогодишните времеви хоризонти, рисковете са реални и изискват активно управление.
Това е последващо допълнение към нашите статии за клиничното валидиране (базата от доказателства преди пускането на пазара, която се разширява от постмаркетинговото наблюдение), как да се напише техническо досие (където се намира планът за PMS в Приложение III), шестмесечната регулаторна пътна карта (къде трябва да се изгради инфраструктурата на PMS) и инженерната реалност на клиничния изкуствен интелект (инфраструктурата за технически мониторинг зад активното наблюдение).
Какво изисква ЕС MDR за следпускане на пазара
Приложение III към РМД на ЕС определя изискванията за следпродажбено наблюдение. Основните задължения:
План за следпродажбено наблюдение. Изготвен преди маркировката CE, описващ как производителят проактивно ще събира и анализира данни за производителността и безопасността на устройството през целия му търговски живот. За устройства с изкуствен интелект планът трябва да разглежда конкретно как ще се наблюдава текущата производителност на системата за изкуствен интелект/машинно обучение.
Доклад за постмаркетингов надзор (за Клас IIa). Годишно обобщение на данните от постмаркетинговия надзор, заключения и всички предприети действия. Докладът включва: обобщение на оплакванията и нежеланите събития, резултати от всяко постмаркетингово клинично проследяване, оценка на съотношението полза-риск и всички заключения относно необходимостта от актуализации на клиничната оценка или досието за управление на риска.
Периодичен доклад за безопасност (PSUR). За изделия от клас IIa, PSUR се подава до нотифицирания орган поне на всеки две години. За клас IIb и III, поне годишно. PSUR обобщава резултатите от постмаркетинговия надзор и заключенията за профила полза-риск на изделието.
Клинично проследяване след пускане на пазара (PMCF). Когато първоначалната клинична оценка има ограничения — ограничен размер на извадката, валидиране на едно място, кратък период на проследяване — производителят трябва да проведе PMCF, за да запълни тези пропуски след пускане на пазара. За устройства с изкуствен интелект, подаващи заявки с ограничени данни от множество места, обикновено се очаква план за PMCF, предлагащ проучвания на ефективността в реални условия.
Докладване за бдителност. Сериозни инциденти (неизправност или влошаване на характеристиките, водещи до смърт на пациент или сериозно влошаване на здравето) трябва да бъдат докладвани на съответния компетентен орган в рамките на определени срокове съгласно член 87 от MDR — 15 дни за сериозни инциденти, 10 дни за смърт или непредвидено сериозно влошаване и 2 дни за сериозни заплахи за общественото здраве. За устройства с изкуствен интелект, сериозен инцидент включва вреда за пациента, пряко дължаща се на фалшиво отрицателен или фалшиво положителен резултат от алгоритъма.
Регистрация в EUDAMED. Изделията и производителите трябва да бъдат регистрирани в EUDAMED, а потоците от данни след пускане на пазара (подаване на PSUR, доклади за сериозни инциденти, коригиращи действия за безопасност на място) се попълват в базата данни. Инфраструктурата на PMS трябва да е съобразена с форматите и сроковете за подаване на данни в EUDAMED.
Проблемът с постпазарния надзор, специфичен за изкуствения интелект: Промяна в дистрибуцията
Производителността на традиционните медицински устройства е стабилна, освен ако физическото устройство не се влоши или не бъде модифицирано. ЕКГ монитор, който работи правилно днес, ще работи правилно и следващата година, ако се поддържа и калибрира.
Производителността на AI модела не е стабилна по този начин. Моделът генерира резултати въз основа на статистически модели, научени от данни за обучение. Ако разпределението на входните данни, които моделът получава в производствения процес, се отклони от разпределението си за обучение, производителността може да се влоши без никаква промяна в самия модел.
Промяната в разпределението на медицинския ИИ се дължи на:
Промени в скенерите и оборудването. Болниците обновяват оборудването за образна диагностика на цикли от 5–10 години. Мамографски изкуствен интелект, валидиран върху изображения от модел скенер X, може да се представя различно върху изображения от модел скенер Y, дори ако и двата вида изображения произвеждат диагностично еквивалентни за рентгенолог. Моделът е научил текстурни и контрастни модели, специфични за обучаващия скенер.
Клиничният работен процес се променя. Ако клиничният протокол за назначаване на ЕКГ се промени – преминавайки от селективно назначаване към по-широк скрининг – съставът на случаите, постъпващи в системата с изкуствен интелект, се променя. Модел, валидиран върху симптоматични пациенти, може да получи по-висок дял асимптоматични пациенти с по-ниска разпространеност на заболяването, което значително променя неговата положителна предсказваща стойност.
Промени в популацията. Демографските данни на пациентите се променят с течение на времето – застаряващо население, миграция, променящи се модели на заболявания. Модел, обучен върху популация от пациенти от 2022 г., може да се сблъска със систематично различни разпределения на входните данни до 2026 г.
Сезонни и времеви вариации. Някои състояния имат сезонни модели (грип, определени респираторни прояви). Модел, обучен върху данни от един период от време, може да види различни разпределения на входните данни по различно време на годината.
Практиката за анотиране се променя. Ако конвенциите за етикетиране на основните данни във вашите данни за обучение отразяват клиничните практически насоки от 2023 г. и тези насоки бъдат актуализирани през 2025 г., прогнозите на модела може систематично да се отклоняват от новия стандарт – не защото моделът се е променил, а защото се е променило определението за основни данни.
Как изглежда активното наблюдение на производителността
За медицинско устройство с изкуствен интелект „мониторингът на оплакванията“ не е достатъчна стратегия за наблюдение след пускането му на пазара. Необходимо е активно наблюдение на производителността, което открива промени в дистрибуцията и влошаване на производителността, преди те да причинят вреда на пациента.
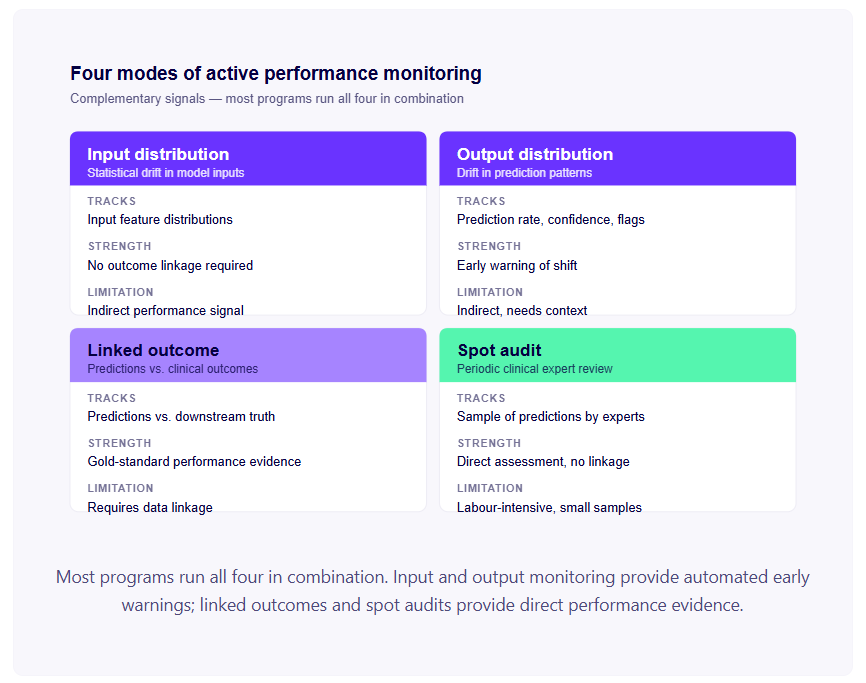
Мониторинг на разпределението на входните данни. Статистическо проследяване на разпределението на входните данни, които моделът получава в производствения процес. За образна диагностика (ИИ): проследяване на разпределенията на показателите за качество на изображението, параметрите на придобиване, демографските данни на пациента. За ЕКГ ИИ: проследяване на разпределенията на сърдечната честота, показателите за качество на сигнала, идентификаторите на оборудването за придобиване. Статистическите тестове (индекс на стабилност на популацията, KL дивергенция, тестове на Колмогоров-Смирнов) могат да открият кога разпределението на входните данни се е изместило значително от разпределението на обучението.
Мониторингът на разпределението на входните данни не изисква данни за клиничните резултати — той следи данните, влизащи в модела, а не коректността на неговите резултати. Това го прави приложим без сложно свързване на клинични данни и осигурява ранен предупредителен сигнал.
Мониторинг на разпределението на изходните данни. Проследяване на разпределението на изходните данни на модела — делът на случаите, маркирани като високорискови, разпределението на оценките за доверие, делът на случаите, маркирани като извън разпределението. Промените в разпределението на изходните данни могат да показват промяна в разпределението на входните данни, промени в клиничната практика или влошаване на производителността на модела.
Свързан мониторинг на резултатите. Когато данните за клиничните резултати могат да бъдат свързани с прогнози на модели — чрез интеграция с електронни здравни карти (ЕЗД), програми за клиничен одит или официални PMCF проучвания — проследяването на връзката между прогнозите на моделите и последващите клинични резултати предоставя директни доказателства за ефективността. Това е златният стандарт за мониторинг на ефективността в реалния свят, но изисква инфраструктура за свързване на данни и подходящо управление за достъп до данните за резултатите.
Точков одит. Периодичен клиничен преглед на произволна извадка от прогнози на модели, с клинична експертна оценка дали прогнозите са били правилни. Трудоемък, но предоставя директни доказателства за ефективността, без да се изисква автоматизирано свързване на данни.
Клинично проследяване след пускане на пазара (PMCF): Запълване на празнотите в доказателствата
PMCF е планирано, систематично събиране на клинични данни след пускането им на пазара, за да се запълнят празнините в доказателствената база от първоначалната клинична оценка. За медицински изделия с изкуствен интелект PMCF обикновено се изисква, когато:
- Първоначалната валидация беше на едно място и са необходими данни за ефективността на множество места.
- Наборът от данни за валидиране имаше ограничено представителство на специфични демографски групи
- Периодът на проследяване във валидационното проучване е бил недостатъчен за оценка на дългосрочното представяне.
- Клиничната среда, в която се използва устройството, се различава от средата за валидиране
Планът за PMCF (представен като част от техническото досие за маркировката CE) описва планираните проучвания, техните цели, методология и срокове за завършване. Докладът за оценка на PMCF (актуализиран периодично, обикновено ежегодно) обобщава резултатите и техните последици за профила полза-риск на изделието.
За устройства с изкуствен интелект, често срещани PMCF дизайни:
Многоцентрово проучване на производителността. Разгръщане на устройството в допълнителни клинични центрове, които не са включени в първоначалното валидиране, и събиране на проспективни данни за производителността. Основната цел обикновено е да се потвърди, че производителността в центровете за валидиране е обобщена за по-широк диапазон от клинични среди.
Дългосрочно кохортно проследяване. Проследяване на пациенти, чиито случаи са били оценени от системата за изкуствен интелект (ИИ), и проследяване на клиничните резултати за продължителен период от време. Особено важно за диагностичния ИИ, където клиничните последици от фалшиво отрицателните резултати могат да станат очевидни едва месеци или години по-късно.
Специфични подгрупови проучвания. Целенасочено събиране на данни за демографски групи, недостатъчно представени в първоначалната валидация — специфични възрастови групи, етнически произход, клинични съпътстващи заболявания.
Управление на актуализациите на моделите: Най-трудният проблем след пускането на пазара
Всеки производител на медицински изделия с изкуствен интелект в крайна сметка се сблъсква с въпроса: искаме да подобрим нашия модел. Какво изисква това съгласно EU MDR?
Рамката на ЕС за MDR не е предназначена за итеративно подобряване на модела на машинно обучение. Изискванията за управление на промените за нови устройства произтичат от приложение II/III на MDR (поддържане на техническа документация), член 10 (задължения на производителя) и член 56 (повторна оценка от нотифициран орган), както и от контрол на софтуерните промени по ISO 13485 и IEC 62304. Заедно те изискват оценка дали промяна в устройство с маркировка CE представлява „съществена промяна“, изискваща нова оценка на съответствието.
Какво представлява значителна промяна за медицинско изделие с изкуствен интелект? EU MDR не предоставя ясен отговор. MDCG 2020-3 — първоначално насоки за значителни промени съгласно преходните разпоредби на член 120 за остарели сертифицирани по MDD устройства — предоставя определения, които са широко приети като фактическа рамка. За устройствата с изкуствен интелект въпросът се решава постепенно чрез практиката на нотифицираните органи и нововъзникващите насоки, но към 2026 г. няма ясен консенсус.
Практическите подходи:
Документирана процедура за оценка на промените. Определете предварително критерии за класифициране на актуализациите на модела като незначителни (подобрение на производителността в рамките на обхвата, без промяна в предвидената употреба, поддържане на валидирана производителност), умерени (промяна в обхвата на данните за обучение, промяна в архитектурата на модела, добавени нови клинични състояния към обхвата) или значителни (промяна в предвидената употреба, съществена промяна в архитектурата, значителна промяна в производителността). Помолете нотифицирания орган да прегледа и приеме тази процедура като част от първоначалното ви техническо досие.
Подходът на предварително определения план за контрол на промените (PCCP). Формализиран от FDA през декември 2024 г. за подавания в САЩ, все по-често споменаван в регулаторните дискусии в ЕС. Предварително посочете видовете актуализации на модела, които очаквате, методологията за валидиране, която ще приложите, и критериите за ефективност, които потвърждават, че актуализацията е безопасна. Получете одобрение от нотифициран орган на PCCP като част от първоначалното си подаване. След това внедрете актуализации в рамките на предварително одобрения обхват без нова пълна оценка на съответствието.
Консервативен подход за големи актуализации. За промени извън предварително определения обхват — ново предназначение, нови клинични показания, фундаментални промени в архитектурата — планирайте оценка на значителните промени, която може да изисква преглед от нотифициран орган и може да доведе до нова оценка на съответствието. Това са важни етапи от разработването на продукта, а не рутинна поддръжка на модела.
Законът на ЕС за изкуствения интелект (ИИ) – постмаркетингово наслагване
За медицинските изделия с изкуствен интелект, обвързани с ЕС, Законът за изкуствен интелект добавя задължения след пускане на пазара в допълнение към рамката за управление на риска (PMS) на MDR. От август 2026 г., с влезлите в сила разпоредби на Закона за изкуствен интелект за високорискови медицински изделия, тези изисквания се прилагат за всяко медицинско изделие от клас IIa+, което включва система с изкуствен интелект.
Съответните задължения след пускането на пазара съгласно Закона за изкуствения интелект:
Система за мониторинг след пускане на пазара (член 72). Документирана система за събиране, анализ и предприемане на действия въз основа на данни за производителността на системата с ИИ през целия ѝ жизнен цикъл. Обхватът се припокрива с MDR PMS, но е очертан конкретно около характеристиките на системата с ИИ — пристрастност, производителност в различните подгрупи, прозрачност за внедрителите, ефективност на човешкия надзор.
Докладване на сериозни инциденти (член 73). Законът за изкуствения интелект има свои собствени изисквания за докладване на сериозни инциденти, свързани със системи с изкуствен интелект, като сроковете и правомощията на получателите могат да се различават от докладването за бдителност по MDR. За медицинските изделия с изкуствен интелект от клас IIa и двата режима се прилагат паралелно.
Задействащи фактори за съществени модификации (член 43). Законът за изкуствения интелект определя съществени модификации, които задействат нова оценка на съответствието за компонента с изкуствен интелект — припокриващи се, но не идентични с критериите за значителни промени по РМИ.
Мониторинг на точността, устойчивостта и отклоненията през жизнения цикъл (член 15). Текуща документация, че системата с ИИ продължава да отговаря на праговете за точност, устойчивост и отклонения, спрямо които е била оценена при оценяването на съответствието.
Практическото значение: програмите, които вече работят по MDR PMS, могат да интегрират постепенно задълженията за постмаркетинг по Закона за изкуствения интелект. Новите програми трябва да планират паралелно документацията за постмаркетинг и на двата режима — отделни работни потоци за отчитане, но до голяма степен споделена базова инфраструктура от данни.
Мониторинг на киберсигурността след пускането на пазара
Освен бдителността относно MDR, мрежовите медицински устройства с изкуствен интелект са изправени пред текущи задължения за киберсигурност съгласно насоките на IEC 81001-5-1 и MDCG 2019-16. Работният поток по киберсигурност след пускане на пазара включва:
- Мониторинг на уязвимости (проследяване на CVE, съвети за сигурност за зависимости)
- Реагиране при инциденти със сигурността (ограничаване, комуникация, регулаторно докладване, където е приложимо)
- Управлението и разпространението на корекции са съобразени с процеса ви за контрол на промените в софтуера
- Периодична преоценка на заплахите с развитието на средата за внедряване
За медицински устройства с изкуствен интелект, хоствани в облак, мониторингът на киберсигурността след пускането им на пазара е непрекъснат — повърхността на заплахите се променя постоянно. Планирайте отделен работен поток за наблюдение на киберсигурността, наред с работния поток за клинична PMS.
Инфраструктурата за следпазарно наблюдение, от която се нуждаете преди маркировката CE
Изграждането на инфраструктура за постмаркетингов надзор след поставянето на маркировката CE, под натиска на първия краен срок за съответствие, е значително по-трудно от изграждането ѝ като част от архитектурата на устройството. Минималната инфраструктура, която трябва да е налице при поставянето на маркировката CE:
Структурирано регистриране. Всеки извод за модела се регистрира с: времева маркировка, идентификатор на входните данни (анонимизиран), използвана версия на модела, изход (прогноза и достоверност) и всички повдигнати флагове за качество. Съхранява се във формат, който може да се извлече, достъпен за вашия екип за следпродажбено наблюдение.
Процес на приемане на жалби. Формален механизъм за получаване, записване и проследяване на жалби от клинични потребители, с определени срокове за реакция и критерии за ескалация на потенциални сериозни инциденти.
Процедура за бдителност. Документиран процес за оценка на потенциални сериозни инциденти, определяне на необходимостта от докладване съгласно член 87 от Регламента на ЕС за медицинските препарати (MDR) и член 73 от Закона на ЕС за изкуствения интелект (AI Act) и своевременно докладване до съответните компетентни органи.
Механизъм за събиране на данни за PMCF. Ако вашият план за PMCF включва събиране на данни за проспективни резултати, механизмът за събиране на данни – работен процес за съгласие на пациента, подход за свързване на данни, съхранение и управление – трябва да е в действие в момента на маркировката CE, а не да е проектиран след това.
Табла за мониторинг на производителността. Мониторингът на разпределението на входните и изходните данни, откриването на аномалии и задействането на предупреждения при достигане на прагове трябва да са оперативни от първия ден на клиничното внедряване. Преоборудването на мониторинга, след като производителността вече се е влошила, е твърде късно.
Следпазарното наблюдение е работен поток, който повечето програми за сърдечен ИИ и медицински ИИ изграждат последни и за който плащат първи. Ресурсното му осигуряване като паралелен работен поток преди маркировката CE, а не след това, е единственото най-ефективно следпазарно решение.
Където се вписва Vector Labs
Работим с производители на медицински изделия с изкуствен интелект по отношение на постмаркетинговия надзор в три аспекта: проектиране на инфраструктурата на PMS преди пускане на пазара (регистриране, табла за мониторинг, работни процеси за оплаквания и бдителност, събиране на данни от PMCF); текуща работа на PMS (изготвяне на доклад за PMS и PSUR, откриване на промени в дистрибуцията, решения за управление на промените); и постмаркетингова интеграция на Закона за ИИ/MDR (системи за мониторинг по член 72, докладване на инциденти по член 73, поддържане на съответствието със Закона за ИИ).
Ако обмисляте работния поток PMS за медицинско устройство с изкуствен интелект — или се сблъсквате с въпроси относно актуализирането на модела след пускането му на пазара — свържете се с vector-labs.ai .
За по-широката серия: класификацията SaMD обхваща дали изобщо е необходимо техническо досие; шестмесечната регулаторна пътна карта обхваща къде в програмата трябва да бъде вградена PMS инфраструктура; клиничното валидиране обхваща базата от доказателства преди пускането на пазара; как да се напише техническо досие обхваща структурата на документацията по Приложение II и Приложение III; инженерната реалност на клиничния ИИ обхваща инфраструктурата за технически мониторинг.
Често задавани въпроси
Докладът за надзор след пускане на пазара (PMS Report) представлява вътрешно обобщение на данните, заключенията и действията, свързани с надзора след пускане на пазара — той трябва да бъде на разположение за проверка, но не се подава рутинно. Докладът за периодично актуализиране на информацията за безопасността (PSUR) е по-изчерпателен документ, който се подава до вашия нотифициран орган и обобщава резултатите от надзора след пускане на пазара, както и профила на ползите и рисковете на изделието. За изделия от клас IIa PSUR се изисква най-малко на всеки две години; за изделия от клас IIb и III — най-малко веднъж годишно.
Значителни промени — такива, които засягат безопасността, експлоатационните характеристики, предназначението или основната конструкция на устройството — обикновено изискват участието на нотифициран орган. За AI/ML значителните промени обикновено включват промени в предназначението, разширяване към нови клинични показания, фундаментални промени в архитектурата на модела или промяна в характеристиките на материала. Преобучението в рамките на обхвата на нови данни, корекциите на праговете и незначителните подобрения в характеристиките обикновено не изискват нова оценка на съответствието, ако са обхванати от предварително определен план за контрол на промените, приет от вашия нотифициран орган.
Три често срещани подхода. Първо, интеграция с електронни здравни досиета (EHR), при която обектите, където се прилага продуктът, споделят анонимизирани данни за резултатите с производителя съгласно подходящи споразумения за обмен на данни. Второ, официални проучвания за наблюдение след пускане на пазара (PMCF), при които проспективно се събират данни за резултатите от извадка от случаи. Трето, програми за клиничен одит, при които клиничните партньори извършват периодичен преглед на медицинските досиета и споделят обобщени данни за ефективността. Всеки от тях изисква предварителна договорна уредба и организационна работа, които трябва да бъдат планирани преди получаването на маркировката „CE“.
Като минимум: структурирано записване на всеки извод на модела (времева отметка, идентификатор на входните данни, версия на модела, резултат, индикатори за качество); процедура за приемане на жалби с определени срокове за отговор; процедура за бдителност, която документира начина, по който се оценяват и докладват сериозните инциденти; както и механизъм за събиране на данни от проучването за постмаркетингово наблюдение (PMCF), ако вашият план включва проспективно събиране на данни за резултатите. Въвеждането на тези елементи след получаване на маркировката „СЕ“, при натиска на времето, свързан с първия краен срок за докладване, е значително по-трудно, отколкото тяхното вграждане като част от архитектурата на устройството.
Законът за изкуствения интелект въвежда допълнителни изисквания към Регламента за медицинските изделия (MDR) за системи с изкуствен интелект с висока степен на риск (включително медицински изделия с изкуствен интелект от клас IIa+ съгласно MDR). Член 72 изисква документирана система за наблюдение след пускане на пазара, специфична за ИИ; член 73 изисква докладване на сериозни инциденти в сроковете, определени в закона; член 43 определя съществените модификации, които изискват нова оценка на съответствието за ИИ компонента. Рамките на Закона за ИИ и MDR след пускане на пазара се припокриват, но изискват отделни работни процеси за документиране и докладване.
FDA официално утвърди PCCP през декември 2024 г. Регулаторната рамка на ЕС се приближава към сходни принципи, но все още няма официален еквивалент на PCCP съгласно MDR. Практическият подход: проектирайте вашия PCCP така, че да отговаря на изискванията на FDA, документирайте същия план за управление на промените във вашия технически файл по MDR (в рамките на контрола на промените в софтуера по IEC 62304 и плана за PMS) и поискайте вашият нотифициран орган да прегледа и приеме обхвата. Резултатът е функционално еквивалентен и в двете юрисдикции.
Съгласно член 2, параграф 65 от Регламента на ЕС за медицинските изделия (MDR) сериозен инцидент е всяка неизправност или влошаване на характеристиките или експлоатационните качества на изделие, което е довело, пряко или косвено, до смърт, сериозно влошаване на здравето или сериозна заплаха за общественото здраве. За медицинските изделия с изкуствен интелект това включва вреда за пациента, пряко свързана с фалшив отрицателен резултат (пропусната диагноза, довела до вреда), фалшив положителен резултат (ненужна интервенция с неблагоприятни последици) или системно влошаване на работата, засягащо множество пациенти. Срокове за докладване съгласно член 87: 15 дни за общи сериозни инциденти, 10 дни за смърт или неочаквано сериозно влошаване на здравето, 2 дни за сериозни заплахи за общественото здраве.
Планът се актуализира непрекъснато като динамичен документ. Докладът за системата за мониторинг на безопасността (PMS) се изготвя ежегодно за изделия от клас IIa. Докладът за проследяване на безопасността след пускане на пазара (PSUR) се актуализира най-малко на всеки две години за изделия от клас IIa и ежегодно за изделия от клас IIb и III. Значителни промени в нормативната уредба, в данните за реалното функциониране или в самото изделие налагат актуализации извън предвидения график.